22.5 Aromatic Reactions
Learning Objectives
By the end of this section, you will be able to:
- Recognize and predict aromatic reactions such as substitution, oxidation and reduction (or hydrogenation).
Electrophilic Aromatic Substitution
Before seeing how electrophilic aromatic substitutions occur, let’s briefly recall about electrophilic alkene additions. When a reagent such as HCl adds to an alkene, the electrophilic hydrogen ion approaches the π electrons of the double bond and forms a bond to one carbon, leaving a positive charge at the other carbon. This carbocation intermediate then reacts with the nucleophilic Cl– ion to yield the addition product as shown in Figure 22.5a.
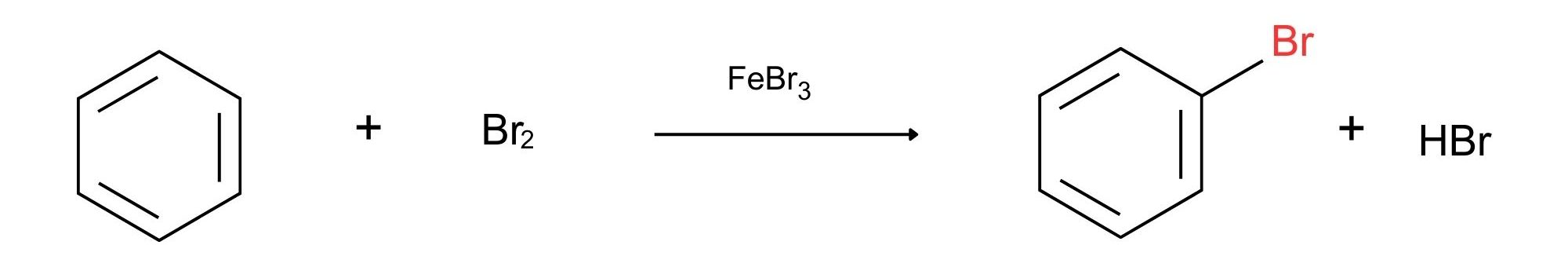
An electrophilic aromatic substitution reaction begins in a similar way, but there are a number of differences. One difference is that aromatic rings are less reactive toward electrophiles than alkenes. For example, Br2 in CH2Cl2 solution reacts instantly with most alkenes but does not react with benzene at room temperature. For bromination of benzene to take place, a catalyst such as FeBr3 is needed. The catalyst makes the Br2 molecule more electrophilic by polarizing it to give a FeBr4–Br+ species that reacts as if it were Br+. The polarized Br2 molecule then reacts with the nucleophilic benzene ring to yield a nonaromatic carbocation intermediate that is doubly allylic and has three resonance forms as shown in Figure 22.5b.
Although more stable than a typical alkyl carbocation because of resonance, the intermediate in electrophilic aromatic substitution is nevertheless much less stable than the starting benzene ring itself, with its 150 kJ/mol (36 kcal/mol) of aromatic stability. Thus, the reaction of an electrophile with a benzene ring is endergonic, has a substantial activation energy, and is rather slow. Figure 22.5c. shows an energy diagram comparing the reaction of an electrophile with an alkene and with benzene. The benzene reaction is slower (higher ∆G‡) because the starting material is more stable. Additionally, the benzene reaction is slower than the alkene reaction because of the stability of the aromatic ring.
Another difference between alkene addition and aromatic substitution occurs after the carbocation intermediate has formed. Instead of adding Br– to give an addition product, the carbocation intermediate loses H+ from the bromine-bearing carbon to give a substitution product. The net effect of reaction of Br2 with benzene is the substitution of H+ by Br+ by the overall mechanism shown in Figure 22.5d. The reaction occurs in two steps and involves a resonance-stabilized carbocation intermediate.
In summary, electrophilic aromatic substitution of benzene with HBr using FeBr3 as a catalyst yields bromobenzene (Figure 22.5e.).
Watch Intro to Electrophilic Aromatic Substitution: Crash Course Organic Chemistry #37 (youtube.com) (12 min).
Video Source: Crash Course. (2021, October 13). Intro to Electrophilic Aromatic Substitution: Crash Course Organic Chemistry #37 (youtube.com) [Video]. YouTube.
Aromatic Halogenation
Chlorine and iodine can be introduced into aromatic rings by electrophilic substitution reactions just as bromine can, but fluorine is too reactive and only poor yields of monofluoroaromatic products are obtained by direct fluorination. Instead, other sources of “F+” are used, in which a fluorine atom is bonded to a positively charged nitrogen. One of the most common such reagents goes by the acronym F-TEDA-BF4 in the presence of trifluoromethanesulfonic acid (TfOH). (You don’t need to know the full name of F-TEDA, which is sold under the name Selectfluor.). An example in Figure 22.5f. shows the electrophilic substitution of toluene using F-TEDA-BF4 as a catalyst to yield o-fluorotoluene and p-fluorotoluene.
More than 20% of all pharmaceutical agents sold contain fluorine, including 30% of the top 100 drugs sold. Sitagliptin (Januvia), used to treat type 2 diabetes, fluoxetine (Prozac), an antidepressant, and atorvastatin (Lipitor), a statin used to lower cholesterol, are examples (Figure 22.5g.).
Aromatic rings react with Cl2 in the presence of FeCl3 catalyst to yield chlorobenzenes, just as they react with Br2 and FeBr3 (Figure 22.5h.). This kind of reaction is used in the synthesis of numerous pharmaceutical agents, including the antiallergy medication loratadine, marketed as Claritin.
Iodine itself is unreactive toward aromatic rings, so an oxidizing agent such as hydrogen peroxide or a copper salt such as CuCl2 must be added to the reaction. These substances accelerate the iodination reaction by oxidizing I2 to a more powerful electrophilic species that reacts as if it were I+. The aromatic ring then reacts with I+ in the typical way, yielding a substitution product (Figure 22.5i.).
Electrophilic aromatic halogenations also occur in the biosynthesis of many naturally occurring molecules, particularly those produced by marine organisms. In humans, the best-known example occurs in the thyroid gland during the biosynthesis of thyroxine, a hormone involved in regulating growth and metabolism. The amino acid tyrosine is first iodinated by thyroid peroxidase, and two of the iodinated tyrosine molecules then couple (Figure 22.5j.). The electrophilic iodinating agent is an I+ species, perhaps hypoiodous acid (HIO), that is formed from iodide ion by oxidation with H2O2.
Aromatic Nitration
Aromatic rings are nitrated by reaction with a mixture of concentrated nitric and sulfuric acids. The electrophile is the nitronium ion, NO2+, which is formed from HNO3 by protonation and loss of water. The nitronium ion reacts with benzene to yield a carbocation intermediate, and loss of H+ from this intermediate gives the neutral substitution product, nitrobenzene (Figure 22.5k.). The mechanism for electrophilic nitration of an aromatic ring is shown below (Figure 22.5k.).
An electrostatic potential map of the reactive electrophile NO2+ shows that the nitrogen atom is most positive.
Electrophilic nitration of an aromatic ring does not occur in nature but is particularly important in the laboratory because the nitro-substituted product can be reduced by reagents such as iron, tin, or SnCl2 to yield the corresponding arylamine, ArNH2. Attachment of an amino group (–NH2) to an aromatic ring by the two-step nitration/reduction sequence is a key part of the industrial synthesis of many dyes and pharmaceutical agents. For example (Figure 22.5l.), nitrobenzene in the presence of Fe, H3O+ and OH– yields aniline.
Aromatic Sulfonation
Aromatic rings can be sulfonated by reaction with so-called fuming sulfuric acid, a mixture of H2SO4 and SO3 (Figure 22.5m.). The reactive electrophile is either HSO3+ or neutral SO3, depending on reaction conditions, and substitution occurs by the same two-step mechanism seen previously for bromination and nitration. Note, however, that the sulfonation reaction is readily reversible. It can occur either forward or backward, depending on the reaction conditions. Sulfonation is favoured in strong acid, but desulfonation is favoured in hot, dilute aqueous acid.
Aromatic sulfonation does not occur naturally but is widely used in the preparation of dyes and pharmaceutical agents. For example, the sulfa drugs, such as sulfanilamide (Figure 22.5n.), were among the first clinically useful antibiotics. Although largely replaced today by more effective agents, sulfa drugs are still used in the treatment of meningitis and urinary tract infections. These drugs are prepared commercially by a process that involves aromatic sulfonation as its key step.
Aromatic Hydroxylation
Direct hydroxylation of an aromatic ring to yield a hydroxybenzene (a phenol) is difficult and rarely done in the laboratory but occurs much more frequently in biological pathways. An example (Figure 22.5o.) is the hydroxylation of p-hydroxyphenylacetate to give 3,4-dihydroxyphenylacetate. The reaction is catalyzed by p-hydroxyphenylacetate-3-hydroxylase and requires molecular oxygen plus the coenzyme reduced flavin adenine dinucleotide, abbreviated FADH2.
Oxidation of Alkyl Side Chains
Despite its unsaturation, the benzene ring is inert to strong oxidizing agents such as KMnO4, which will cleave alkene carbon–carbon bonds. It turns out, however, that the presence of the aromatic ring has a dramatic effect on the reactivity of alkyl side chains. These side chains react rapidly with oxidizing agents and are converted into carboxyl groups, –CO2H.The net effect is conversion of an alkylbenzene into a benzoic acid, Ar–R→Ar–CO2H. Butylbenzene is oxidized by aqueous KMnO4 to give benzoic acid, for instance (Figure 22.5p.).
A similar oxidation is employed industrially for the preparation of the terephthalic acid used in the production of polyester fibers. Worldwide, approximately 118 million tons per year of terephthalic acid is produced by oxidation of p-xylene, using air as the oxidant and Co(III) salts as catalyst.
The mechanism of side-chain oxidation is complex and involves reaction of C–H bonds at the position next to the aromatic ring to form intermediate benzylic radicals. tert-Butylbenzene has no benzylic hydrogens, however, and is therefore inert (Figure 22.5r.).
Analogous side-chain oxidations occur in various biosynthetic pathways. The neurotransmitter norepinephrine, for instance, is biosynthesized from dopamine by a benzylic hydroxylation reaction. The process is catalyzed by the copper-containing enzyme dopamine β-monooxygenase and occurs by a radical mechanism. A copper–oxygen species in the enzyme first abstracts the pro-R benzylic hydrogen to give a radical, and a hydroxyl is then transferred from copper to carbon.
Catalytic Hydrogenation of Aromatic Rings
Just as aromatic rings are generally inert to oxidation, they’re also inert to catalytic hydrogenation under conditions that reduce typical alkene double bonds. As a result, it’s possible to reduce an alkene double bond selectively in the presence of an aromatic ring. For example (Figure 22.5s.), 4-phenyl-3-buten-2-one is reduced to 4-phenyl-2-butanone using a palladium catalyst at room temperature and atmospheric pressure. Neither the benzene ring nor the ketone carbonyl group is affected.
To hydrogenate an aromatic ring, it’s necessary either to use a platinum catalyst with hydrogen gas at a pressure of several hundred atmospheres or to use a more effective catalyst such as rhodium on carbon. Under these conditions, aromatic rings are converted into cyclohexanes. For example, o-xylene yields 1,2-dimethylcyclohexane, and 4-tert-butylphenol gives 4-tert-butylcyclohexanol as shown in Figure 22.5t.
For a detailed summary of the various reaction pathways that aromatic compounds undergo, refer to infographic 22.5a.
Watch Diazonium Salts & Nucleophilic Aromatic Substitution: Crash Course Organic Chemistry #47 (youtube.com) (14 min). Some aspects of this video may not apply to this text.
Video Source: Crash Course. (2022, March 2). Diazonium Salts & Nucleophilic Aromatic Substitution: Crash Course Organic Chemistry #47 (youtube.com) [Video]. YouTube.
Friedel-Crafts Reactions
Friedel-Crafts reactions involve the acylation of benzene and substituted benzenes. An acyl group is an alkyl group attached to a carbon-oxygen double bond. If “R” represents any alkyl group, then an acyl group has the formula RCO-. Acylation means substituting an acyl group into something – in this case, into a benzene ring. A hydrogen on the ring is replaced by a group like methyl or ethyl and so on.
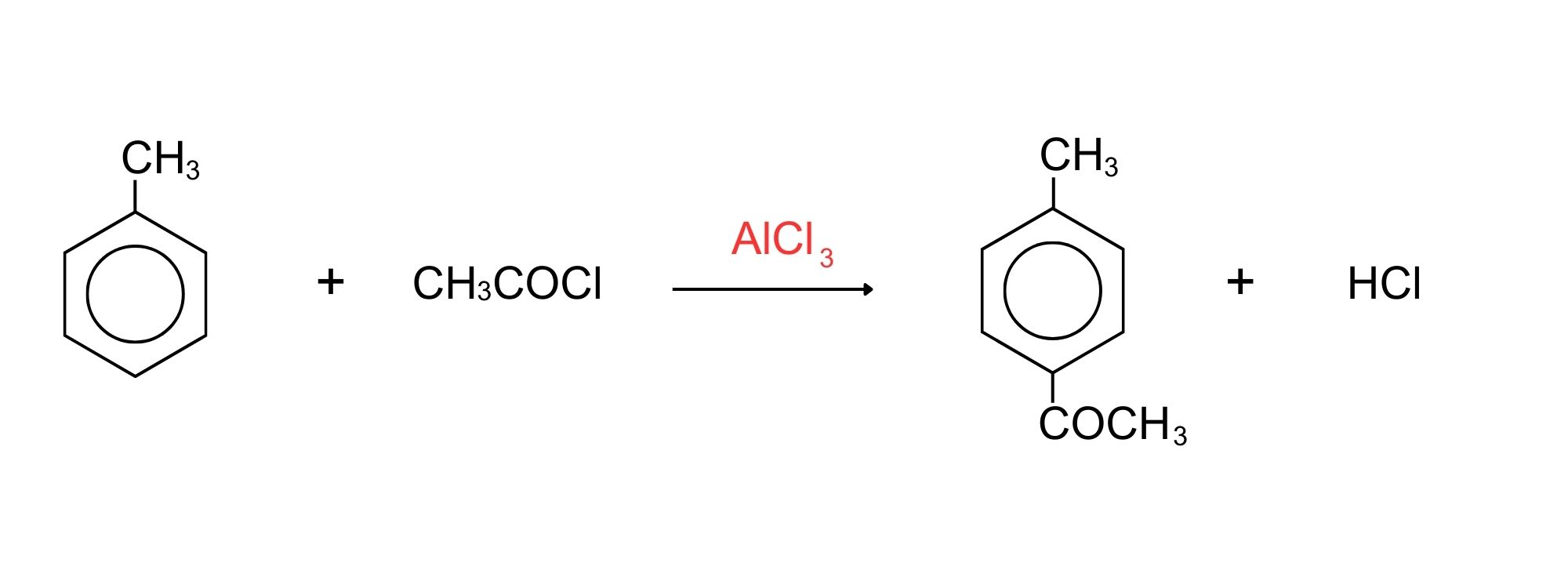
The most commonly used acyl group is CH3CO-. This is called the ethanoyl group, and in this case the reaction is sometimes called “ethanoylation”. In the example which follows we are substituting a CH3CO- group into the ring, but you could equally well use any other acyl group. The most reactive substance containing an acyl group is an acyl chloride (also known as an acid chloride). These have the general formula RCOCl. Benzene is treated with a mixture of ethanoyl chloride, CH3COCl, and aluminium chloride as the catalyst. The mixture is heated to about 60°C for about 30 minutes. A ketone called phenylethanone (old name: acetophenone) is formed as shown in Figure 22.5u.

Note in Figure 22.5u., that the aluminum chloride isn’t written into these equations because it is acting as a catalyst. If you wanted to include it, you could write AlCl3 over the top of the arrow.
The reaction is just the same with methylbenzene except that you have to worry about where the acyl group attaches to the ring relative to the methyl group. Normally, the methyl group in methylbenzene directs new groups into the 2- and 4- positions (assuming the methyl group is in the 1- position). In acylation, though, virtually all the substitution happens in the 4- position (Figure 22.5v.).
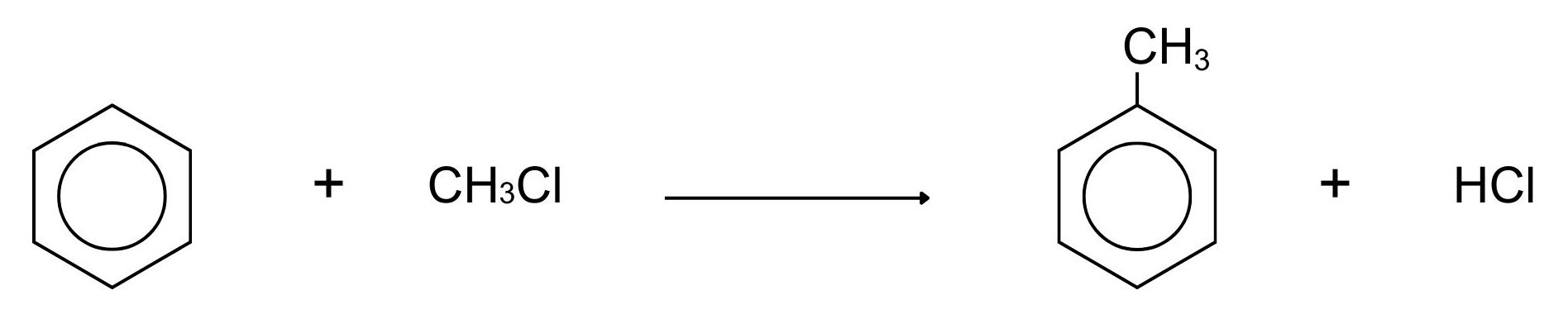
Alkylation means substituting an alkyl group into something – in this case into a benzene ring. A hydrogen on the ring is replaced by a group like methyl or ethyl and so on. Benzene reacts at room temperature with a chloroalkane (for example, chloromethane or chloroethane) in the presence of aluminum chloride as a catalyst. In Figure 22.5w., a methyl group is substituted, but any other alkyl group could be used in the same way. Substituting a methyl group gives methylbenzene.
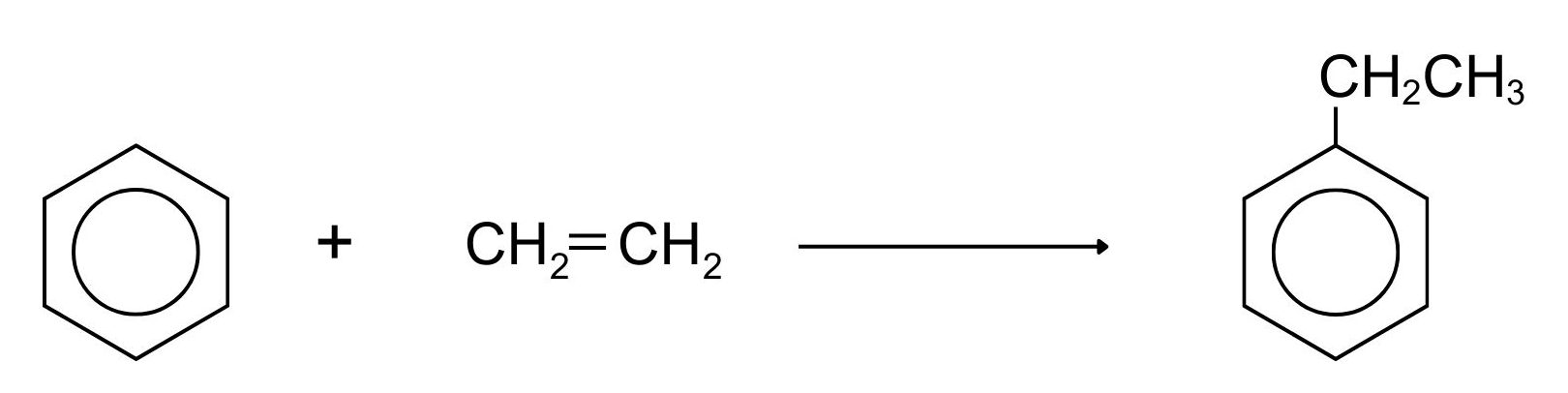
Friedel-Crafts reactions are also involved in industrial processes such as the manufacturing of ethylbenzene (Figure 22.5x.). Ethylbenzene is an important industrial chemical used to make styrene (phenylethene), which in turn is used to make polystyrene – poly(phenylethene). It is manufactured from benzene and ethene. There are several ways of doing this, some of which use a variation on Friedel-Crafts alkylation.
Attribution & References
Except where otherwise noted, this page is adapted by Adrienne Richards and Samantha Sullivan Sauer from:
- “16.1 Electrophilic Aromatic Substitution Reactions: Bromination“, “16.2 Other Aromatic Substitutions“, “16.8 Oxidation of Aromatic Compounds” and “16.9 Reduction of Aromatic Compounds” In Organic Chemistry (OpenStax) by John McMurray, CC BY-NC-SA 4.0. Access for free at Organic Chemistry (OpenStax)
- “Friedel-Crafts Reactions” by Jim Clark In Supplemental Modules (Organic Chemistry) is shared under a CC BY-NC 4.0
an alkyl group attached to a carbon-oxygen double bond
substituting an acyl group into something
