16. Microbial Mechanisms of Pathogenicity
16.3 Virulence Factors of Bacterial and Viral Pathogens
Learning Objectives
- Explain how virulence factors contribute to signs and symptoms of infectious disease
- Differentiate between endotoxins and exotoxins
- Describe and differentiate between various types of exotoxins
- Describe the mechanisms viruses use for adhesion and antigenic variation
In the previous section, we explained that some pathogens are more virulent than others. This is due to the unique virulence factors produced by individual pathogens, which determine the extent and severity of disease they may cause. A pathogen’s virulence factors are encoded by genes that can be identified using molecular Koch’s postulates. When genes encoding virulence factors are inactivated, virulence in the pathogen is diminished. In this section, we examine various types and specific examples of virulence factors and how they contribute to each step of pathogenesis.
Virulence Factors for Adhesion
As discussed in the previous section, the first two steps in pathogenesis are exposure and adhesion. Recall that an adhesin is a protein or glycoprotein found on the surface of a pathogen that attaches to receptors on the host cell. Adhesins are found on bacterial, viral, fungal, and protozoan pathogens. One example of a bacterial adhesin is type 1 fimbrial adhesin, a molecule found on the tips of fimbriae of enterotoxigenic E. coli (ETEC). Recall that fimbriae are hairlike protein bristles on the cell surface. Type 1 fimbrial adhesin allows the fimbriae of ETEC cells to attach to the mannose glycans expressed on intestinal epithelial cells. Table 16.7 lists common adhesins found in some of the pathogens we have discussed or will be seeing later in this chapter.
Table 16.7. Bacterial Adhesins and Their Host Attachment Sites
| Some Bacterial Adhesins and Their Host Attachment Sites | |||
|---|---|---|---|
| Pathogen | Disease | Adhesin | Attachment Site |
| Streptococcus pyogenes | Strep throat | Protein F | Respiratory epithelial cells |
| Streptococcus mutans | Dental caries | Adhesin P1 | Teeth |
| Neisseria gonorrhoeae | Gonorrhea | Type IV pili | Urethral epithelial cells |
| Enterotoxigenic E. coli (ETEC) | Traveler’s diarrhoea | Type 1 fimbriae | Intestinal epithelial cells |
| Vibrio cholerae | Cholera | N-methylphenylalanine pili | Intestinal epithelial cells |
CLINICAL FOCUS: Part 3
The presence of bacteria in Michael’s blood is a sign of infection, since blood is normally sterile. There is no indication that the bacteria entered the blood through an injury. Instead, it appears the portal of entry was the gastrointestinal route. Based on Michael’s symptoms, the results of his blood test, and the fact that Michael was the only one in the family to partake of the hot dogs, the physician suspects that Michael is suffering from a case of listeriosis.
Listeria monocytogenes, the facultative intracellular pathogen that causes listeriosis, is a common contaminant in ready-to-eat foods such as lunch meats and dairy products. Once ingested, these bacteria invade intestinal epithelial cells and translocate to the liver, where they grow inside hepatic cells. Listeriosis is fatal in about one in five normal healthy people, and mortality rates are slightly higher in patients with pre-existing conditions that weaken the immune response. A cluster of virulence genes encoded on a pathogenicity island is responsible for the pathogenicity of L. monocytogenes. These genes are regulated by a transcriptional factor known as peptide chain release factor 1 (PrfA). One of the genes regulated by PrfA is hyl, which encodes a toxin known as listeriolysin O (LLO), which allows the bacterium to escape vacuoles upon entry into a host cell. A second gene regulated by PrfA is actA, which encodes for a surface protein known as actin assembly-inducing protein (ActA). ActA is expressed on the surface of Listeria and polymerizes host actin. This enables the bacterium to produce actin tails, move around the cell’s cytoplasm, and spread from cell to cell without exiting into the extracellular compartment.
Michael’s condition has begun to worsen. He is now experiencing a stiff neck and hemiparesis (weakness of one side of the body). Concerned that the infection is spreading, the physician decides to conduct additional tests to determine what is causing these new symptoms.
- What kind of pathogen causes listeriosis, and what virulence factors contribute to the signs and symptoms Michael is experiencing?
- Is it likely that the infection will spread from Michael’s blood? If so, how might this explain his new symptoms?
Jump to the next Clinical Focus box. Go back to the previous Clinical Focus box.
Bacterial Exoenzymes and Toxins as Virulence Factors
After exposure and adhesion, the next step in pathogenesis is invasion, which can involve enzymes and toxins. Many pathogens achieve invasion by entering the bloodstream, an effective means of dissemination because blood vessels pass close to every cell in the body. The downside of this mechanism of dispersal is that the blood also includes numerous elements of the immune system. Various terms ending in –aemia are used to describe the presence of pathogens in the bloodstream. The presence of bacteria in blood is called bacteraemia. Bacteraemia involving pyogens (pus-forming bacteria) is called pyaemia. When viruses are found in the blood, it is called viraemia. The term toxaemia describes the condition when toxins are found in the blood. If bacteria are both present and multiplying in the blood, this condition is called septicaemia.
Patients with septicaemia are described as septic, which can lead to shock, a life-threatening decrease in blood pressure (systolic pressure <90 mm Hg) that prevents cells and organs from receiving enough oxygen and nutrients. Some bacteria can cause shock through the release of toxins (virulence factors that can cause tissue damage) and lead to low blood pressure. Gram-negative bacteria are engulfed by immune system phagocytes, which then release tumour necrosis factor, a molecule involved in inflammation and fever. Tumour necrosis factor binds to blood capillaries to increase their permeability, allowing fluids to pass out of blood vessels and into tissues, causing swelling, or ooedema (Figure 16.10). With high concentrations of tumour necrosis factor, the inflammatory reaction is severe and enough fluid is lost from the circulatory system that blood pressure decreases to dangerously low levels. This can have dire consequences because the heart, lungs, and kidneys rely on normal blood pressure for proper function; thus, multi-organ failure, shock, and death can occur.
Exoenzymes
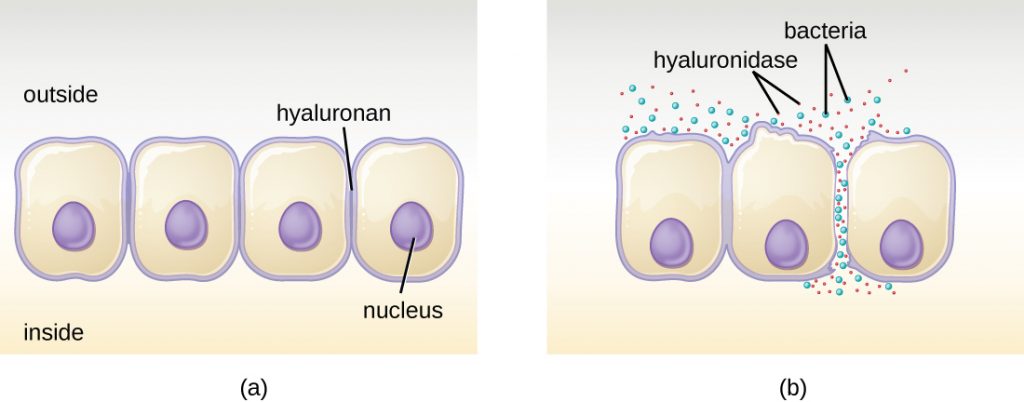
Some pathogens produce extracellular enzymes, or exoenzymes, that enable them to invade host cells and deeper tissues. Exoenzymes have a wide variety of targets. Some general classes of exoenzymes and associated pathogens are listed in Table 16.8. Each of these exoenzymes functions in the context of a particular tissue structure to facilitate invasion or support its own growth and defend against the immune system. For example, hyaluronidase S, an enzyme produced by pathogens like Staphylococcus aureus, Streptococcus pyogenes, and Clostridium perfringens, degrades the glycoside hylauronan (hyaluronic acid), which acts as an intercellular cement between adjacent cells in connective tissue (Figure 16.11). This allows the pathogen to pass through the tissue layers at the portal of entry and disseminate elsewhere in the body (Figure 16.11).
Table 16.8. Classes of Exoenzymes and Their Targets
| Some Classes of Exoenzymes and Their Targets | ||
|---|---|---|
| Class | Example | Function |
| Glycohydrolases | Hyaluronidase S in Staphylococcus aureus | Degrades hyaluronic acid that cements cells together to promote spreading through tissues |
| Nucleases | DNAse produced by S. aureus | Degrades DNA released by dying cells (bacteria and host cells) that can trap the bacteria, thus promoting spread |
| Phospholipases | Phospholipase C of Bacillus anthracis | Degrades phospholipid bilayer of host cells, causing cellular lysis, and degrade membrane of phagosomes to enable escape into the cytoplasm |
| Proteases | Collagenase in Clostridium perfringens | Degrades collagen in connective tissue to promote spread |
Pathogen-produced nucleases, such as DNAse produced by S. aureus, degrade extracellular DNA as a means of escape and spreading through tissue. As bacterial and host cells die at the site of infection, they lyse and release their intracellular contents. The DNA chromosome is the largest of the intracellular molecules, and masses of extracellular DNA can trap bacteria and prevent their spread. S. aureus produces a DNAse to degrade the mesh of extracellular DNA so it can escape and spread to adjacent tissues. This strategy is also used by S. aureus and other pathogens to degrade and escape webs of extracellular DNA produced by immune system phagocytes to trap the bacteria.
Enzymes that degrade the phospholipids of cell membranes are called phospholipases. Their actions are specific in regard to the type of phospholipids they act upon and where they enzymatically cleave the molecules. The pathogen responsible for anthrax, B. anthracis, produces phospholipase C. When B. anthracis is ingested by phagocytic cells of the immune system, phospholipase C degrades the membrane of the phagosome before it can fuse with the lysosome, allowing the pathogen to escape into the cytoplasm and multiply. Phospholipases can also target the membrane that encloses the phagosome within phagocytic cells. As described earlier in this chapter, this is the mechanism used by intracellular pathogens such as L. monocytogenes and Rickettsia to escape the phagosome and multiply within the cytoplasm of phagocytic cells. The role of phospholipases in bacterial virulence is not restricted to phagosomal escape. Many pathogens produce phospholipases that act to degrade cell membranes and cause lysis of target cells. These phospholipases are involved in lysis of red blood cells, white blood cells, and tissue cells.
Bacterial pathogens also produce various protein-digesting enzymes, or proteases. Proteases can be classified according to their substrate target (e.g., serine proteases target proteins with the amino acid serine) or if they contain metals in their active site (e.g., zinc metalloproteases contain a zinc ion, which is necessary for enzymatic activity).
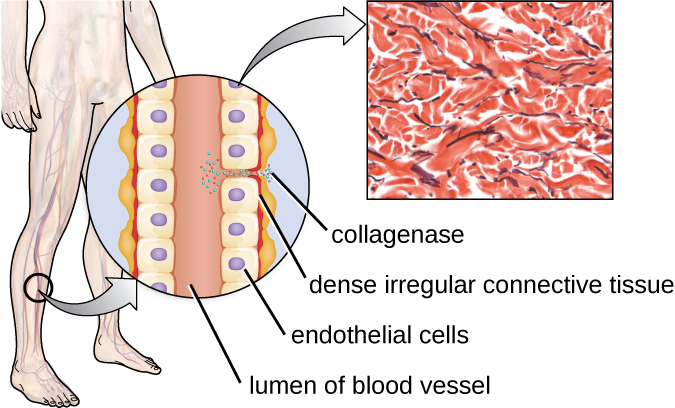
One example of a protease that contains a metal ion is the exoenzyme collagenase. Collagenase digests collagen, the dominant protein in connective tissue. Collagen can be found in the extracellular matrix, especially near mucosal membranes, blood vessels, nerves, and in the layers of the skin. Similar to hyaluronidase, collagenase allows the pathogen to penetrate and spread through the host tissue by digesting this connective tissue protein. The collagenase produced by the gram-positive bacterium Clostridium perfringens, for example, allows the bacterium to make its way through the tissue layers and subsequently enter and multiply in the blood (septicaemia). C. perfringens then uses toxins and a phospholipase to cause cellular lysis and necrosis. Once the host cells have died, the bacterium produces gas by fermenting the muscle carbohydrates. The widespread necrosis of tissue and accompanying gas are characteristic of the condition known as gas gangrene (Figure 16.12).

Two types of cell death are apoptosis and necrosis. Visit this website to learn more about the differences between these mechanisms of cell death and their causes.
Toxins
In addition to exoenzymes, certain pathogens are able to produce toxins, biological poisons that assist in their ability to invade and cause damage to tissues. The ability of a pathogen to produce toxins to cause damage to host cells is called toxigenicity.
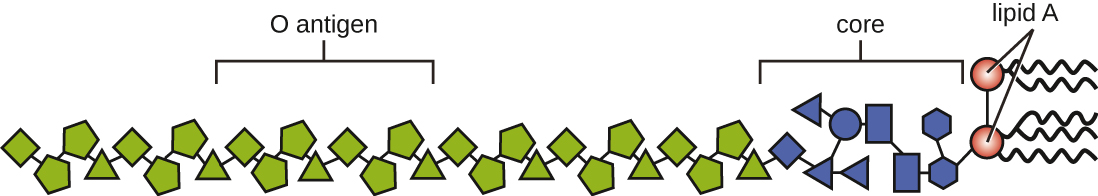
Toxins can be categorized as endotoxins or exotoxins. The lipopolysaccharide (LPS) found on the outer membrane of gram-negative bacteria is called endotoxin (Figure 16.13). During infection and disease, gram-negative bacterial pathogens release endotoxin either when the cell dies, resulting in the disintegration of the membrane, or when the bacterium undergoes binary fission. The lipid component of endotoxin, lipid A, is responsible for the toxic properties of the LPS molecule. Lipid A is relatively conserved across different genera of gram-negative bacteria; therefore, the toxic properties of lipid A are similar regardless of the gram-negative pathogen. In a manner similar to that of tumour necrosis factor, lipid A triggers the immune system’s inflammatory response (seeInflammation and Fever). If the concentration of endotoxin in the body is low, the inflammatory response may provide the host an effective defence against infection; on the other hand, high concentrations of endotoxin in the blood can cause an excessive inflammatory response, leading to a severe drop in blood pressure, multi-organ failure, and death.
A classic method of detecting endotoxin is by using the Limulus amoebocyte lysate (LAL) test. In this procedure, the blood cells (amoebocytes) of the horseshoe crab (Limulus polyphemus) is mixed with a patient’s serum. The amoebocytes will react to the presence of any endotoxin. This reaction can be observed either chromogenically (colour) or by looking for coagulation (clotting reaction) to occur within the serum. An alternative method that has been used is an enzyme-linked immunosorbent assay (ELISA) that uses antibodies to detect the presence of endotoxin.
Unlike the toxic lipid A of endotoxin, exotoxins are protein molecules that are produced by a wide variety of living pathogenic bacteria. Although some gram-negative pathogens produce exotoxins, the majority are produced by gram-positive pathogens. Exotoxins differ from endotoxin in several other key characteristics, summarized in Table 16.9. In contrast to endotoxin, which stimulates a general systemic inflammatory response when released, exotoxins are much more specific in their action and the cells they interact with. Each exotoxin targets specific receptors on specific cells and damages those cells through unique molecular mechanisms. Endotoxin remains stable at high temperatures, and requires heating at 121 °C (250 °F) for 45 minutes to inactivate. By contrast, most exotoxins are heat labile because of their protein structure, and many are denatured (inactivated) at temperatures above 41 °C (106 °F). As discussed earlier, endotoxin can stimulate a lethal inflammatory response at very high concentrations and has a measured LD50 of 0.24 mg/kg. By contrast, very small concentrations of exotoxins can be lethal. For example, botulinum toxin, which causes botulism, has an LD50 of 0.000001 mg/kg (240,000 times more lethal than endotoxin).
Table 16.9. Comparison of Endotoxin and Bacterial Exotoxins
| Comparison of Endotoxin and Exotoxins Produced by Bacteria | ||
|---|---|---|
| Characteristic | Endotoxin | Exotoxin |
| Source | Gram-negative bacteria | Gram-positive (primarily) and gram-negative bacteria |
| Composition | Lipid A component of lipopolysaccharide | Protein |
| Effect on host | General systemic symptoms of inflammation and fever | Specific damage to cells dependent upon receptor-mediated targeting of cells and specific mechanisms of action |
| Heat stability | Heat stable | Most are heat labile, but some are heat stable |
| LD50 | High | Low |
The exotoxins can be grouped into three categories based on their target: intracellular targeting, membrane disrupting, and superantigens. Table 16.10 provides examples of well-characterized toxins within each of these three categories.
Table 16.10 Common Exotoxins and Associated Bacterial Pathogens
| Some Common Exotoxins and Associated Bacterial Pathogens | |||
|---|---|---|---|
| Category | Example | Pathogen | Mechanism and Disease |
| Intracellular-targeting toxins | Cholera toxin | Vibrio cholerae | Activation of adenylate cyclase in intestinal cells, causing increased levels of cyclic adenosine monophosphate (cAMP) and secretion of fluids and electrolytes out of cell, causing diarrhoea |
| Tetanus toxin | Clostridium tetani | Inhibits the release of inhibitory neurotransmitters in the central nervous system, causing spastic paralysis | |
| Botulinum toxin | Clostridium botulinum | Inhibits release of the neurotransmitter acetylcholine from neurons, resulting in flaccid paralysis | |
| Diphtheria toxin | Corynebacterium diphtheriae | Inhibition of protein synthesis, causing cellular death | |
| Membrane-disrupting toxins | Streptolysin | Streptococcus pyogenes | Proteins that assemble into pores in cell membranes, disrupting their function and killing the cell |
| Pneumolysin | Streptococcus pneumoniae | ||
| Alpha-toxin | Staphylococcus aureus | ||
| Alpha-toxin | Clostridium perfringens | Phospholipases that degrade cell membrane phospholipids, disrupting membrane function and killing the cell | |
| Phospholipase C | Pseudomonas aeruginosa | ||
| Beta-toxin | Staphylococcus aureus | ||
| Superantigens | Toxic shock syndrome toxin | Staphylococcus aureus | Stimulates excessive activation of immune system cells and release of cytokines (chemical mediators) from immune system cells. Life-threatening fever, inflammation, and shock are the result. |
| Streptococcal mitogenic exotoxin | Streptococcus pyogenes | ||
| Streptococcal pyrogenic toxins | Streptococcus pyogenes | ||
The intracellular targeting toxins comprise two components: A for activity and B for binding. Thus, these types of toxins are known as A-B exotoxins (Figure 16.14). The B component is responsible for the cellular specificity of the toxin and mediates the initial attachment of the toxin to specific cell surface receptors. Once the A-B toxin binds to the host cell, it is brought into the cell by endocytosis and entrapped in a vacuole. The A and B subunits separate as the vacuole acidifies. The A subunit then enters the cell cytoplasm and interferes with the specific internal cellular function that it targets.
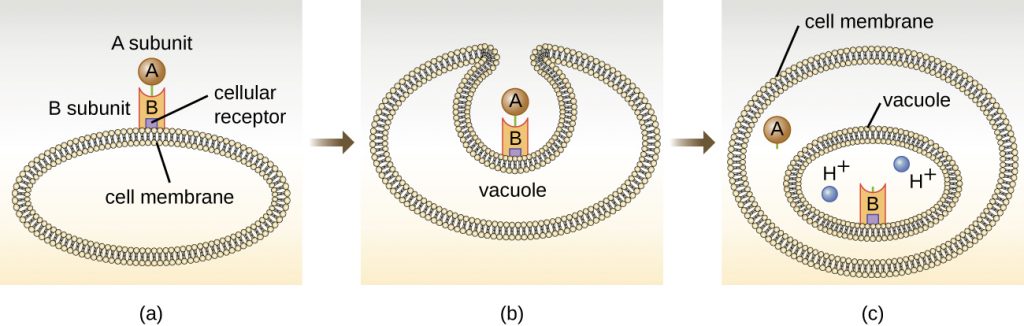
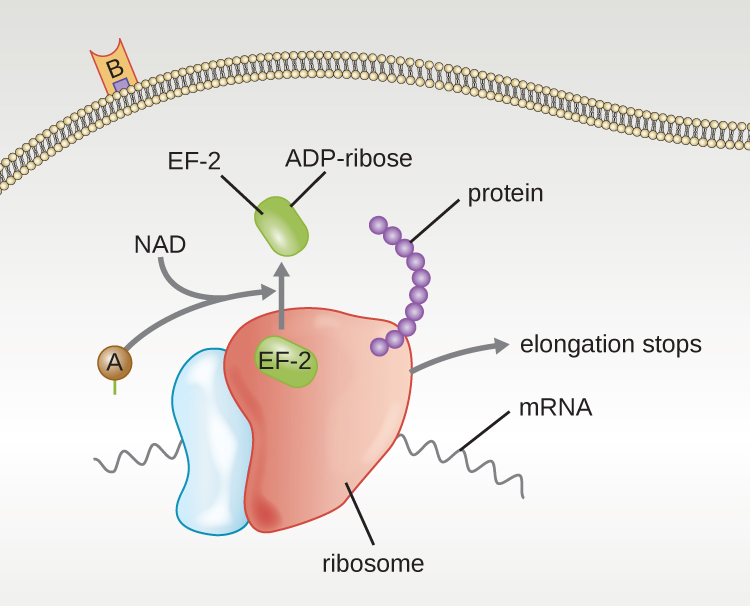
Four unique examples of A-B toxins are the diphtheria, cholera, botulinum, and tetanus toxins. The diphtheria toxin is produced by the gram-positive bacterium Corynebacterium diphtheriae, the causative agent of nasopharyngeal and cutaneous diphtheria. After the A subunit of the diphtheria toxin separates and gains access to the cytoplasm, it facilitates the transfer of adenosine diphosphate (ADP)-ribose onto an elongation-factor protein (EF-2) that is needed for protein synthesis. Hence, diphtheria toxin inhibits protein synthesis in the host cell, ultimately killing the cell (Figure 16.15).
Cholera toxin is an enterotoxin produced by the gram-negative bacterium Vibrio cholerae and is composed of one A subunit and five B subunits. The mechanism of action of the cholera toxin is complex. The B subunits bind to receptors on the intestinal epithelial cell of the small intestine. After gaining entry into the cytoplasm of the epithelial cell, the A subunit activates an intracellular G protein. The activated G protein, in turn, leads to the activation of the enzyme adenyl cyclase, which begins to produce an increase in the concentration of cyclic AMP (a secondary messenger molecule). The increased cAMP disrupts the normal physiology of the intestinal epithelial cells and causes them to secrete excessive amounts of fluid and electrolytes into the lumen of the intestinal tract, resulting in severe “rice-water stool” diarrhoea characteristic of cholera.
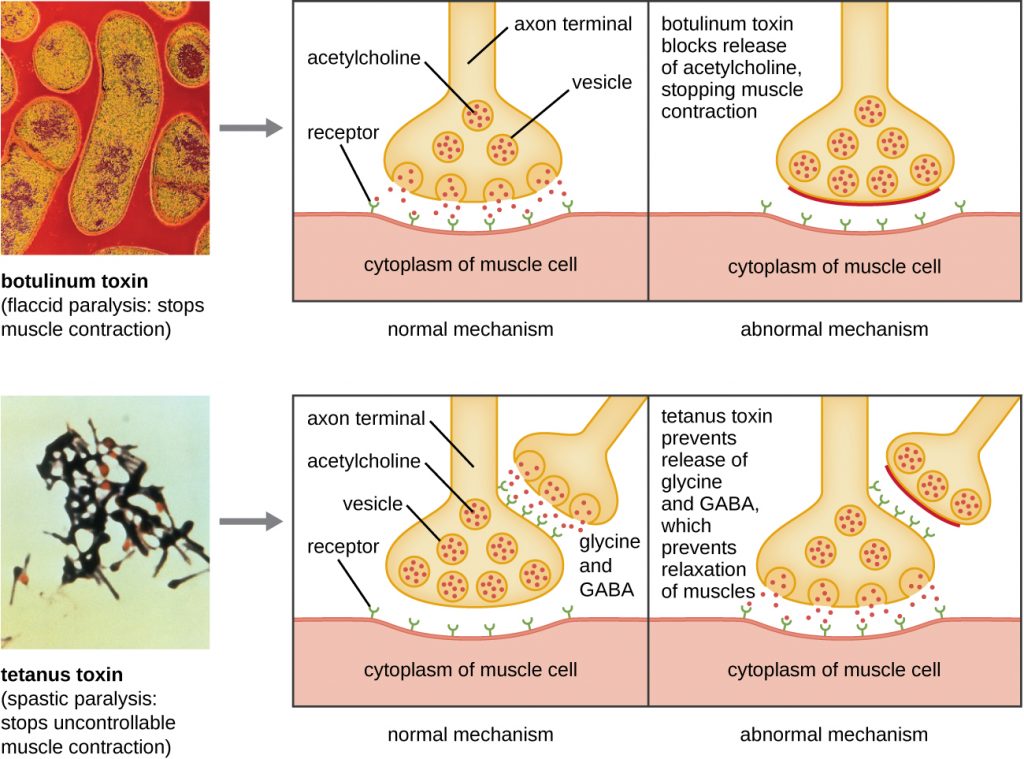
Botulinum toxin (also known as botox) is a neurotoxin produced by the gram-positive bacterium Clostridium botulinum. It is the most acutely toxic substance known to date. The toxin is composed of a light A subunit and heavy protein chain B subunit. The B subunit binds to neurons to allow botulinum toxin to enter the neurons at the neuromuscular junction. The A subunit acts as a protease, cleaving proteins involved in the neuron’s release of acetylcholine, a neurotransmitter molecule. Normally, neurons release acetylcholine to induce muscle fibre contractions. The toxin’s ability to block acetylcholine release results in the inhibition of muscle contractions, leading to muscle relaxation. This has the potential to stop breathing and cause death. Because of its action, low concentrations of botox are used for cosmetic and medical procedures, including the removal of wrinkles and treatment of overactive bladder.
Click this link to see an animation of how the cholera toxin functions. This link provides an animation of how the botulinum toxin functions.
Another neurotoxin is tetanus toxin, which is produced by the gram-positive bacterium Clostridium tetani. This toxin also has a light A subunit and heavy protein chain B subunit. Unlike botulinum toxin, tetanus toxin binds to inhibitory interneurons, which are responsible for release of the inhibitory neurotransmitters glycine and gamma-aminobutyric acid (GABA). Normally, these neurotransmitters bind to neurons at the neuromuscular junction, resulting in the inhibition of acetylcholine release. Tetanus toxin inhibits the release of glycine and GABA from the interneuron, resulting in permanent muscle contraction. The first symptom is typically stiffness of the jaw (lockjaw). Violent muscle spasms in other parts of the body follow, typically culminating with respiratory failure and death. Figure 16.16 shows the actions of both botulinum and tetanus toxins.

Membrane-disrupting toxins affect cell membrane function either by forming pores or by disrupting the phospholipid bilayer in host cell membranes. Two types of membrane-disrupting exotoxins are haemolysins and leukocidins, which form pores in cell membranes, causing leakage of the cytoplasmic contents and cell lysis. These toxins were originally thought to target red blood cells (erythrocytes) and white blood cells (leukocytes), respectively, but we now know they can affect other cells as well. The gram-positive bacterium Streptococcus pyogenes produces streptolysins, water-soluble haemolysins that bind to the cholesterol moieties in the host cell membrane to form a pore. The two types of streptolysins, O and S, are categorized by their ability to cause hemolysis in erythrocytes in the absence or presence of oxygen. Streptolysin O is not active in the presence of oxygen, whereas streptolysin S is active in the presence of oxygen. Other important pore-forming membrane-disrupting toxins include alpha toxin of Staphylococcus aureus and pneumolysin of Streptococcus pneumoniae.
Bacterial phospholipases are membrane-disrupting toxins that degrade the phospholipid bilayer of cell membranes rather than forming pores. We have already discussed the phospholipases associated with B. anthracis, L. pneumophila, and Rickettsia species that enable these bacteria to effect the lysis of phagosomes. These same phospholipases are also haemolysins. Other phospholipases that function as haemolysins include the alpha toxin of Clostridium perfringens, phospholipase C of P. aeruginosa, and beta toxin of Staphylococcus aureus.
Some strains of S. aureus also produce a leukocidin called Panton-Valentine leukocidin (PVL). PVL consists of two subunits, S and F. The S component acts like the B subunit of an A-B exotoxin in that it binds to glycolipids on the outer plasma membrane of animal cells. The F-component acts like the A subunit of an A-B exotoxin and carries the enzymatic activity. The toxin inserts and assembles into a pore in the membrane. Genes that encode PVL are more frequently present in S. aureus strains that cause skin infections and pneumonia.[1] PVL promotes skin infections by causing oedema, erythema (reddening of the skin due to blood vessel dilation), and skin necrosis. PVL has also been shown to cause necrotizing pneumonia. PVL promotes pro-inflammatory and cytotoxic effects on alveolar leukocytes. This results in the release of enzymes from the leukocytes, which, in turn, cause damage to lung tissue.
The third class of exotoxins is the superantigens. These are exotoxins that trigger an excessive, nonspecific stimulation of immune cells to secrete cytokines (chemical messengers). The excessive production of cytokines, often called a cytokine storm, elicits a strong immune and inflammatory response that can cause life-threatening high fevers, low blood pressure, multi-organ failure, shock, and death. The prototype superantigen is the toxic shock syndrome toxin of S. aureus. Most toxic shock syndrome cases are associated with vaginal colonization by toxin-producing S. aureus in menstruating women; however, colonization of other body sites can also occur. Some strains of Streptococcus pyogenes also produce superantigens; they are referred to as the streptococcal mitogenic exotoxins and the streptococcal pyrogenic toxins.
- Describe how exoenzymes contribute to bacterial invasion.
- Explain the difference between exotoxins and endotoxin.
- Name the three classes of exotoxins.
Virulence Factors for Survival in the Host and Immune Evasion
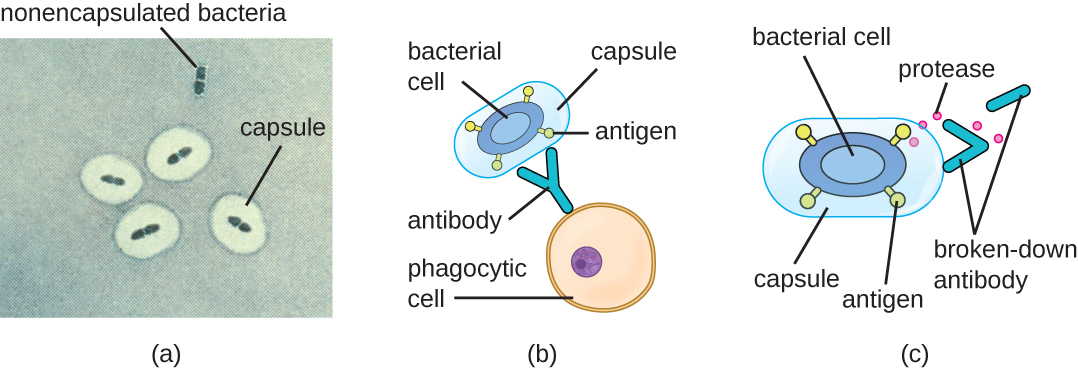
Evading the immune system is also important to invasiveness. Bacteria use a variety of virulence factors to evade phagocytosis by cells of the immune system. For example, many bacteria produce capsules, which are used in adhesion but also aid in immune evasion by preventing ingestion by phagocytes. The composition of the capsule prevents immune cells from being able to adhere and then phagocytose the cell. In addition, the capsule makes the bacterial cell much larger, making it harder for immune cells to engulf the pathogen (Figure 16.17). A notable capsule-producing bacterium is the gram-positive pathogen Streptococcus pneumoniae, which causes pneumococcal pneumonia, meningitis, septicaemia, and other respiratory tract infections. Encapsulated strains of S. pneumoniae are more virulent than nonencapsulated strains and are more likely to invade the bloodstream and cause septicaemia and meningitis.
Some pathogens can also produce proteases to protect themselves against phagocytosis. As described in Adaptive Specific Host Defences, the human immune system produces antibodies that bind to surface molecules found on specific bacteria (e.g., capsules, fimbriae, flagella, LPS). This binding initiates phagocytosis and other mechanisms of antibacterial killing and clearance. Proteases combat antibody-mediated killing and clearance by attacking and digesting the antibody molecules (Figure 16.17).
In addition to capsules and proteases, some bacterial pathogens produce other virulence factors that allow them to evade the immune system. The fimbriae of certain species of Streptococcus contain M protein, which alters the surface of Streptococcus and inhibits phagocytosis by blocking the binding of the complement molecules that assist phagocytes in ingesting bacterial pathogens. The acid-fast bacterium Mycobacterium tuberculosis (the causative agent of tuberculosis) produces a waxy substance known as mycolic acid in its cell envelope. When it is engulfed by phagocytes in the lung, the protective mycolic acid coat enables the bacterium to resist some of the killing mechanisms within the phagolysosome.
Some bacteria produce virulence factors that promote infection by exploiting molecules naturally produced by the host. For example, most strains of Staphylococcus aureus produce the exoenzyme coagulase, which exploits the natural mechanism of blood clotting to evade the immune system. Normally, blood clotting is triggered in response to blood vessel damage; platelets begin to plug the clot, and a cascade of reactions occurs in which fibrinogen, a soluble protein made by the liver, is cleaved into fibrin. Fibrin is an insoluble, thread-like protein that binds to blood platelets, cross-links, and contracts to form a mesh of clumped platelets and red blood cells. The resulting clot prevents further loss of blood from the damaged blood vessels. However, if bacteria release coagulase into the bloodstream, the fibrinogen-to-fibrin cascade is triggered in the absence of blood vessel damage. The resulting clot coats the bacteria in fibrin, protecting the bacteria from exposure to phagocytic immune cells circulating in the bloodstream.
Whereas coagulase causes blood to clot, kinases have the opposite effect by triggering the conversion of plasminogen to plasmin, which is involved in the digestion of fibrin clots. By digesting a clot, kinases allow pathogens trapped in the clot to escape and spread, similar to the way that collagenase, hyaluronidase, and DNAse facilitate the spread of infection. Examples of kinases include staphylokinases and streptokinases, produced by Staphylococcus aureus and Streptococcus pyogenes, respectively. It is intriguing that S. aureus can produce both coagulase to promote clotting and staphylokinase to stimulate the digestion of clots. The action of the coagulase provides an important protective barrier from the immune system, but when nutrient supplies are diminished or other conditions signal a need for the pathogen to escape and spread, the production of staphylokinase can initiate this process.
A final mechanism that pathogens can use to protect themselves against the immune system is called antigenic variation, which is the alteration of surface proteins so that a pathogen is no longer recognized by the host’s immune system. For example, the bacterium Borrelia burgdorferi, the causative agent of Lyme disease, contains a surface lipoprotein known as VlsE. Because of genetic recombination during DNA replication and repair, this bacterial protein undergoes antigenic variation. Each time fever occurs, the VlsE protein in B. burgdorferi can differ so much that antibodies against previous VlsE sequences are not effective. It is believed that this variation in the VlsE contributes to the ability B. burgdorferi to cause chronic disease. Another important human bacterial pathogen that uses antigenic variation to avoid the immune system is Neisseria gonorrhoeae, which causes the sexually transmitted disease gonorrhoea. This bacterium is well known for its ability to undergo antigenic variation of its type IV pili to avoid immune defences.
- Name at least two ways that a capsule provides protection from the immune system.
- Besides capsules, name two other virulence factors used by bacteria to evade the immune system.
CLINICAL FOCUS: Resolution
Based on Michael’s reported symptoms of stiff neck and hemiparesis, the physician suspects that the infection may have spread to his nervous system. The physician decides to order a spinal tap to look for any bacteria that may have invaded the meninges and cerebrospinal fluid (CSF), which would normally be sterile. To perform the spinal tap, Michael’s lower back is swabbed with an iodine antiseptic and then covered with a sterile sheet. The needle is aseptically removed from the manufacturer’s sealed plastic packaging by the clinician’s gloved hands. The needle is inserted and a small volume of fluid is drawn into an attached sample tube. The tube is removed, capped and a prepared label with Michael’s data is affixed to it. This STAT (urgent or immediate analysis required) specimen is divided into three separate sterile tubes, each with 1 mL of CSF. These tubes are immediately taken to the hospital’s lab, where they are analyzed in the clinical chemistry, haematology, and microbiology departments. The preliminary results from all three departments indicate there is a cerebrospinal infection occurring, with the microbiology department reporting the presence of a gram-positive rod in Michael’s CSF.
These results confirm what his physician had suspected: Michael’s new symptoms are the result of meningitis, acute inflammation of the membranes that protect the brain and spinal cord. Because meningitis can be life threatening and because the first antibiotic therapy was not effective in preventing the spread of infection, Michael is prescribed an aggressive course of two antibiotics, ampicillin and gentamicin, to be delivered intravenously. Michael remains in the hospital for several days for supportive care and for observation. After a week, he is allowed to return home for bed rest and oral antibiotics. After 3 weeks of this treatment, he makes a full recovery.
Go back to the previous Clinical Focus box.
Viral Virulence
Although viral pathogens are not similar to bacterial pathogens in terms of structure, some of the properties that contribute to their virulence are similar. Viruses use adhesins to facilitate adhesion to host cells, and certain enveloped viruses rely on antigenic variation to avoid the host immune defences. These virulence factors are discussed in more detail in the following sections.
Viral Adhesins
One of the first steps in any viral infection is adhesion of the virus to specific receptors on the surface of cells. This process is mediated by adhesins that are part of the viral capsid or membrane envelope. The interaction of viral adhesins with specific cell receptors defines the tropism (preferential targeting) of viruses for specific cells, tissues, and organs in the body. The spike protein hemagglutinin found on Influenzavirus is an example of a viral adhesin; it allows the virus to bind to the sialic acid on the membrane of host respiratory and intestinal cells. Another viral adhesin is the glycoprotein gp20, found on HIV. For HIV to infect cells of the immune system, it must interact with two receptors on the surface of cells. The first interaction involves binding between gp120 and the CD4 cellular marker that is found on some essential immune system cells. However, before viral entry into the cell can occur, a second interaction between gp120 and one of two chemokine receptors (CCR5 and CXCR4) must occur. Table 16.11 lists the adhesins for some common viral pathogens and the specific sites to which these adhesins allow viruses to attach.
Table 16.11. Viral Adhesins and Their Host Attachment Sites
| Some Viral Adhesins and Their Host Attachment Sites | |||
|---|---|---|---|
| Pathogen | Disease | Adhesin | Attachment Site |
| Influenzavirus | Influenza | Hemagglutinin | Sialic acid of respiratory and intestinal cells |
| Herpes simplex virus I or II | Oral herpes, genital herpes | Glycoproteins gB, gC, gD | Heparan sulphate on mucosal surfaces of the mouth and genitals |
| Human immunodeficiency virus | HIV/AIDS | Glycoprotein gp120 | CD4 and CCR5 or CXCR4 of immune system cells |
Antigenic Variation in Viruses
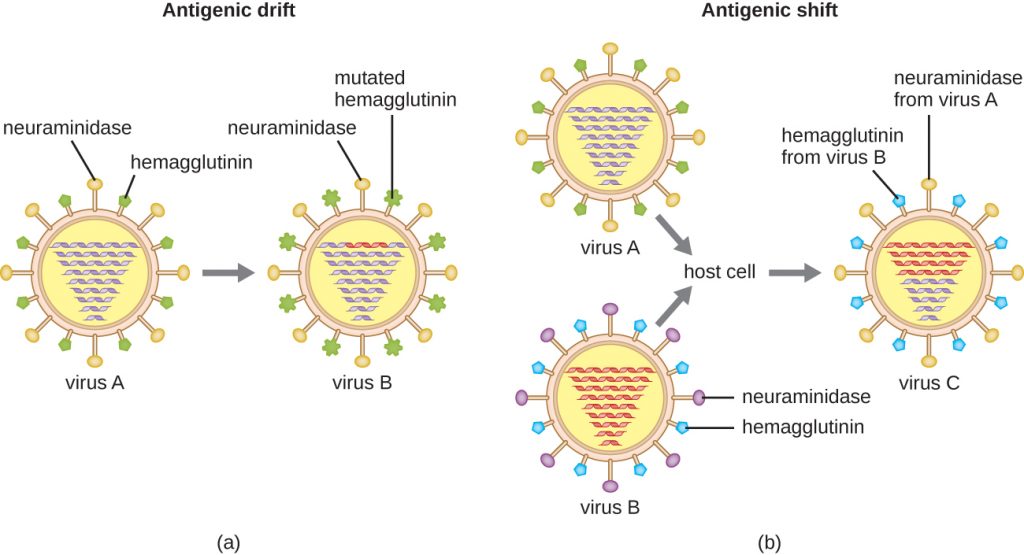
Antigenic variation also occurs in certain types of enveloped viruses, including influenza viruses, which exhibit two forms of antigenic variation: antigenic drift and antigenic shift (Figure 16.18). Antigenic drift is the result of point mutations causing slight changes in the spike proteins hemagglutinin (H) and neuraminidase (N). On the other hand, antigenic shift is a major change in spike proteins due to gene reassortment. This reassortment for antigenic shift occurs typically when two different influenza viruses infect the same host.
The rate of antigenic variation in influenza viruses is very high, making it difficult for the immune system to recognize the many different strains of Influenza virus. Although the body may develop immunity to one strain through natural exposure or vaccination, antigenic variation results in the continual emergence of new strains that the immune system will not recognize. This is the main reason that vaccines against Influenza virus must be given annually. Each year’s influenza vaccine provides protection against the most prevalent strains for that year, but new or different strains may be more prevalent the following year.
- Describe the role of adhesins in viral tropism.
- Explain the difference between antigenic drift and antigenic shift.
Key Takeaways
- Virulence factors contribute to a pathogen’s ability to cause disease.
- Exoenzymes and toxins allow pathogens to invade host tissue and cause tissue damage. Exoenzymes are classified according to the macromolecule they target and exotoxins are classified based on their mechanism of action.
- Bacterial toxins include endotoxin and exotoxins. Endotoxin is the lipid A component of the LPS of the gram-negative cell envelope. Exotoxins are proteins secreted mainly by gram-positive bacteria, but also are secreted by gram-negative bacteria.
- Bacterial pathogens may evade the host immune response by producing capsules to avoid phagocytosis, surviving the intracellular environment of phagocytes, degrading antibodies, or through antigenic variation.
- Viral pathogens use adhesins for initiating infections and antigenic variation to avoid immune defences.
- Influenza viruses use both antigenic drift and antigenic shift to avoid being recognized by the immune system.
Multiple Choice
Fill in the Blank
Critical Thinking
- Two types of toxins are haemolysins and leukocidins. (a) How are these toxins similar? (b) How do they differ?
- Imagine that a mutation in the gene encoding the cholera toxin was made. This mutation affects the A-subunit, preventing it from interacting with any host protein. (a) Would the toxin be able to enter into the intestinal epithelial cell? (b) Would the toxin be able to cause diarrhoea?
Media Attributions
- OSC_Microbio_15_03_hyaluronan
- OSC_Microbio_15_03_CollClost
- microbiology sign © Nick Youngson
- OSC_Microbio_15_03_LPS
- OSC_Microbio_15_03_ABToxin
- OSC_Microbio_15_03_Diphtheria
- OSC_Microbio_15_03_Neurotoxin
- OSC_Microbio_15_03_Phago
- OSC_Microbio_15_03_Flu
- V. Meka. “Panton-Valentine Leukocidin.” http://www.antimicrobe.org/h04c.files/history/PVL-S-aureus.asp ↵
