15. Antimicrobial Drugs
15.3 Mechanisms of Antibacterial Drugs
Learning Objective
- Describe the mechanisms of action associated with drugs that inhibit cell wall biosynthesis, protein synthesis, membrane function, nucleic acid synthesis, and metabolic pathways
An important quality for an antimicrobial drug is selective toxicity, meaning that it selectively kills or inhibits the growth of microbial targets while causing minimal or no harm to the host. Most antimicrobial drugs currently in clinical use are antibacterial because the prokaryotic cell provides a greater variety of unique targets for selective toxicity, in comparison to fungi, parasites, and viruses. Each class of antibacterial drugs has a unique mode of action (the way in which a drug affects microbes at the cellular level), and these are summarized in Figure 15.9 and Table 15.1.
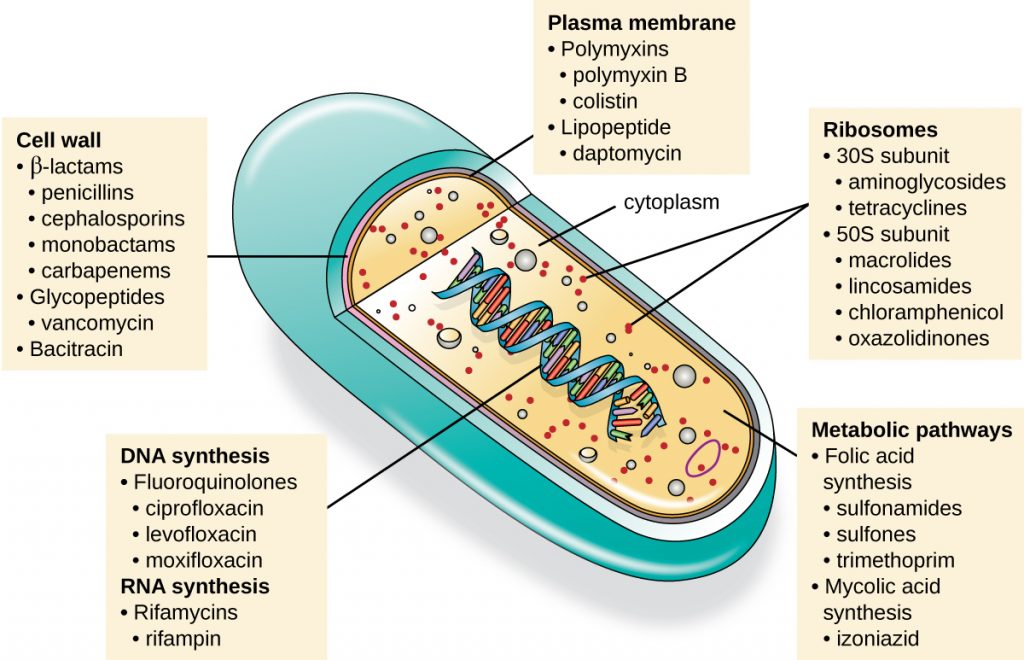
Table 15.1. Common Antibacterial Drugs by Mode of Action
| Common Antibacterial Drugs by Mode of Action | ||
|---|---|---|
| Mode of Action | Target | Drug Class |
| Inhibit cell wall biosynthesis | Penicillin-binding proteins | β-lactams: penicillins, cephalosporins, monobactams, carbapenems |
| Peptidoglycan subunits | Glycopeptides | |
| Peptidoglycan subunit transport | Bacitracin | |
| Inhibit biosynthesis of proteins | 30S ribosomal subunit | Aminoglycosides, tetracyclines |
| 50S ribosomal subunit | Macrolides, lincosamides, chloramphenicol, oxazolidinones | |
| Disrupt membranes | Lipopolysaccharide, inner and outer membranes | Polymyxin B, colistin, daptomycin |
| Inhibit nucleic acid synthesis | RNA | Rifamycin |
| DNA | Fluoroquinolones | |
| Antimetabolites | Folic acid synthesis enzyme | Sulfonamides, trimethoprim |
| Mycolic acid synthesis enzyme | Isonicotinic acid hydrazide | |
| Mycobacterial adenosine triphosphate (ATP) synthase inhibitor | Mycobacterial ATP synthase | Diarylquinoline |
Inhibitors of Cell Wall Biosynthesis
Several different classes of antibacterials block steps in the biosynthesis of peptidoglycan, making cells more susceptible to osmotic lysis (Table 15.2). Therefore, antibacterials that target cell wall biosynthesis are bactericidal in their action. Because human cells do not make peptidoglycan, this mode of action is an excellent example of selective toxicity.
Penicillin, the first antibiotic discovered, is one of several antibacterials within a class called β-lactams. This group of compounds includes the penicillins, cephalosporins, monobactams, and carbapenems, and is characterized by the presence of a β-lactam ring found within the central structure of the drug molecule (Figure 15.10). The β-lactam antibacterials block the crosslinking of peptide chains during the biosynthesis of new peptidoglycan in the bacterial cell wall. They are able to block this process because the β-lactam structure is similar to the structure of the peptidoglycan subunit component that is recognized by the crosslinking transpeptidase enzyme, also known as a penicillin-binding protein (PBP). Although the β-lactam ring must remain unchanged for these drugs to retain their antibacterial activity, strategic chemical changes to the R groups have allowed for development of a wide variety of semisynthetic β-lactam drugs with increased potency, expanded spectrum of activity, and longer half-lives for better dosing, among other characteristics.
Penicillin G and penicillin V are natural antibiotics from fungi and are primarily active against gram-positive bacterial pathogens, and a few gram-negative bacterial pathogens such as Pasteurella multocida. Figure 15.10 summarizes the semisynthetic development of some of the penicillins. Adding an amino group (-NH2) to penicillin G created the aminopenicillins (i.e., ampicillin and amoxicillin) that have increased spectrum of activity against more gram-negative pathogens. Furthermore, the addition of a hydroxyl group (-OH) to amoxicillin increased acid stability, which allows for improved oral absorption. Methicillin is a semisynthetic penicillin that was developed to address the spread of enzymes (penicillinases) that were inactivating the other penicillins. Changing the R group of penicillin G to the more bulky dimethoxyphenyl group provided protection of the β-lactam ring from enzymatic destruction by penicillinases, giving us the first penicillinase-resistant penicillin.
Similar to the penicillins, cephalosporins contain a β-lactam ring (Figure 15.10) and block the transpeptidase activity of penicillin-binding proteins. However, the β-lactam ring of cephalosporins is fused to a six-member ring, rather than the five-member ring found in penicillins. This chemical difference provides cephalosporins with an increased resistance to enzymatic inactivation by β-lactamases. The drug cephalosporin C was originally isolated from the fungus Cephalosporium acremonium in the 1950s and has a similar spectrum of activity to that of penicillin against gram-positive bacteria but is active against more gram-negative bacteria than penicillin. Another important structural difference is that cephalosporin C possesses two R groups, compared with just one R group for penicillin, and this provides for greater diversity in chemical alterations and development of semisynthetic cephalosporins. The family of semisynthetic cephalosporins is much larger than the penicillins, and these drugs have been classified into generations based primarily on their spectrum of activity, increasing in spectrum from the narrow-spectrum, first-generation cephalosporins to the broad-spectrum, fourth-generation cephalosporins. A new fifth-generation cephalosporin has been developed that is active against methicillin-resistant Staphylococcus aureus (MRSA).
The carbapenems and monobactams also have a β-lactam ring as part of their core structure, and they inhibit the transpeptidase activity of penicillin-binding proteins. The only monobactam used clinically is aztreonam. It is a narrow-spectrum antibacterial with activity only against gram-negative bacteria. In contrast, the carbapenem family includes a variety of semisynthetic drugs (imipenem, meropenem, and doripenem) that provide very broad-spectrum activity against gram-positive and gram-negative bacterial pathogens.
The drug vancomycin, a member of a class of compounds called the glycopeptides, was discovered in the 1950s as a natural antibiotic from the actinomycete Amycolatopsis orientalis. Similar to the β-lactams, vancomycin inhibits cell wall biosynthesis and is bactericidal. However, in contrast to the β-lactams, the structure of vancomycin is not similar to that of cell-wall peptidoglycan subunits and does not directly inactivate penicillin-binding proteins. Rather, vancomycin is a very large, complex molecule that binds to the end of the peptide chain of cell wall precursors, creating a structural blockage that prevents the cell wall subunits from being incorporated into the growing N-acetylglucosamine and N-acetylmuramic acid (NAM-NAG) backbone of the peptidoglycan structure (transglycosylation). Vancomycin also structurally blocks transpeptidation. Vancomycin is bactericidal against gram-positive bacterial pathogens, but it is not active against gram-negative bacteria because of its inability to penetrate the protective outer membrane.
The drug bacitracin consists of a group of structurally similar peptide antibiotics originally isolated from Bacillus subtilis. Bacitracin blocks the activity of a specific cell-membrane molecule that is responsible for the movement of peptidoglycan precursors from the cytoplasm to the exterior of the cell, ultimately preventing their incorporation into the cell wall. Bacitracin is effective against a wide range of bacteria, including gram-positive organisms found on the skin, such as Staphylococcus and Streptococcus. Although it may be administered orally or intramuscularly in some circumstances, bacitracin has been shown to be nephrotoxic (damaging to the kidneys). Therefore, it is more commonly combined with neomycin and polymyxin in topical ointments such as Neosporin.
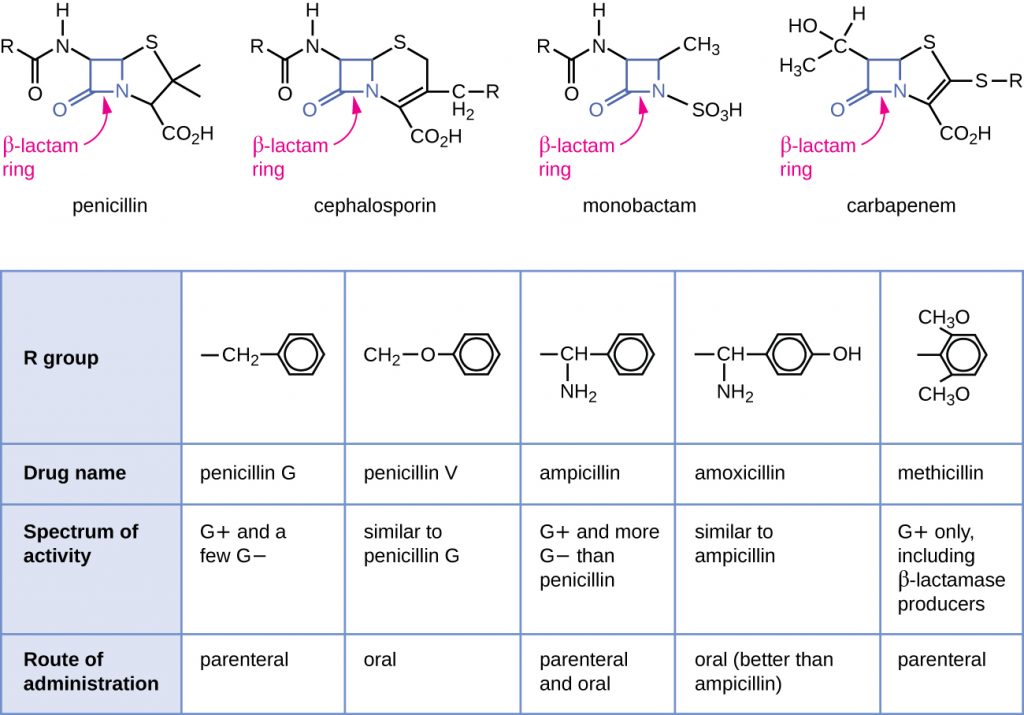
Table 15.2. Drugs that Inhibit Bacterial Cell Wall Synthesis
| Drugs that Inhibit Bacterial Cell Wall Synthesis | ||||
|---|---|---|---|---|
| Mechanism of Action | Drug Class | Specific Drugs | Natural or Semisynthetic | Spectrum of Activity |
| Interact directly with PBPs and inhibit transpeptidase activity | Penicillins | Penicillin G, penicillin V | Natural | Narrow-spectrum against gram-positive and a few gram-negative bacteria |
| Ampicillin, amoxicillin | Semisynthetic | Narrow-spectrum against gram-positive bacteria but with increased gram-negative spectrum | ||
| Methicillin | Semisynthetic | Narrow-spectrum against gram-positive bacteria only, including strains producing penicillinase | ||
| Cephalosporins | Cephalosporin C | Natural | Narrow-spectrum similar to penicillin but with increased gram-negative spectrum | |
| First-generation cephalosporins | Semisynthetic | Narrow-spectrum similar to cephalosporin C | ||
| Second-generation cephalosporins | Semisynthetic | Narrow-spectrum but with increased gram-negative spectrum compared with first generation | ||
| Third- and fourth-generation cephalosporins | Semisynthetic | Broad-spectrum against gram-positive and gram-negative bacteria, including some β-lactamase producers | ||
| Fifth-generation cephalosporins | Semisynthetic | Broad-spectrum against gram-positive and gram-negative bacteria, including MRSA | ||
| Monobactams | Aztreonam | Semisynthetic | Narrow-spectrum against gram-negative bacteria, including some β-lactamase producers | |
| Carbapenems | Imipenem, meropenem, doripenem | Semisynthetic | Broadest spectrum of the β-lactams against gram-positive and gram-negative bacteria, including many β-lactamase producers | |
| Large molecules that bind to the peptide chain of peptidoglycan subunits, blocking transglycosylation and transpeptidation | Glycopeptides | Vancomycin | Natural | Narrow spectrum against gram-positive bacteria only, including multidrug-resistant strains |
| Block transport of peptidoglycan subunits across cytoplasmic membrane | Bacitracin | Bacitracin | Natural | Broad-spectrum against gram-positive and gram-negative bacteria |
- Describe the mode of action of β-lactams.
Inhibitors of Protein Biosynthesis
The cytoplasmic ribosomes found in animal cells (80S) are structurally distinct from those found in bacterial cells (70S), making protein biosynthesis a good selective target for antibacterial drugs. Several types of protein biosynthesis inhibitors are discussed in this section and are summarized in Figure 15.11.
Protein Synthesis Inhibitors That Bind the 30S Subunit
Aminoglycosides are large, highly polar antibacterial drugs that bind to the 30S subunit of bacterial ribosomes, impairing the proofreading ability of the ribosomal complex. This impairment causes mismatches between codons and anticodons, resulting in the production of proteins with incorrect amino acids and shortened proteins that insert into the cytoplasmic membrane. Disruption of the cytoplasmic membrane by the faulty proteins kills the bacterial cells. The aminoglycosides, which include drugs such as streptomycin, gentamicin, neomycin, and kanamycin, are potent broad-spectrum antibacterials. However, aminoglycosides have been shown to be nephrotoxic (damaging to kidney), neurotoxic (damaging to the nervous system), and ototoxic (damaging to the ear).
Another class of antibacterial compounds that bind to the 30S subunit is the tetracyclines. In contrast to aminoglycosides, these drugs are bacteriostatic and inhibit protein synthesis by blocking the association of tRNAs with the ribosome during translation. Naturally occurring tetracyclines produced by various strains of Streptomyces were first discovered in the 1940s, and several semisynthetic tetracyclines, including doxycycline and tigecycline have also been produced. Although the tetracyclines are broad spectrum in their coverage of bacterial pathogens, side effects that can limit their use include phototoxicity, permanent discolouration of developing teeth, and liver toxicity with high doses or in patients with kidney impairment.
Protein Synthesis Inhibitors That Bind the 50S Subunit
There are several classes of antibacterial drugs that work through binding to the 50S subunit of bacterial ribosomes. The macrolide antibacterial drugs have a large, complex ring structure and are part of a larger class of naturally produced secondary metabolites called polyketides, complex compounds produced in a stepwise fashion through the repeated addition of two-carbon units by a mechanism similar to that used for fatty acid synthesis. Macrolides are broad-spectrum, bacteriostatic drugs that block elongation of proteins by inhibiting peptide bond formation between specific combinations of amino acids. The first macrolide was erythromycin. It was isolated in 1952 from Streptomyces erythreus and prevents translocation. Semisynthetic macrolides include azithromycin and telithromycin. Compared with erythromycin, azithromycin has a broader spectrum of activity, fewer side effects, and a significantly longer half-life (1.5 hours for erythromycin versus 68 hours for azithromycin) that allows for once-daily dosing and a short 3-day course of therapy (i.e., Zpac formulation) for most infections. Telithromycin is the first semisynthetic within the class known as ketolides. Although telithromycin shows increased potency and activity against macrolide-resistant pathogens, the US Food and Drug Administration (FDA) has limited its use to treatment of community-acquired pneumonia and requires the strongest “black box warning” label for the drug because of serious hepatotoxicity.
The lincosamides include the naturally produced lincomycin and semisynthetic clindamycin. Although structurally distinct from macrolides, lincosamides are similar in their mode of action to the macrolides through binding to the 50S ribosomal subunit and preventing peptide bond formation. Lincosamides are particularly active against streptococcal and staphylococcal infections.
The drug chloramphenicol represents yet another structurally distinct class of antibacterials that also bind to the 50S ribosome, inhibiting peptide bond formation. Chloramphenicol, produced by Streptomyces venezuelae, was discovered in 1947; in 1949, it became the first broad-spectrum antibiotic that was approved by the FDA. Although it is a natural antibiotic, it is also easily synthesized and was the first antibacterial drug synthetically mass produced. As a result of its mass production, broad-spectrum coverage, and ability to penetrate into tissues efficiently, chloramphenicol was historically used to treat a wide range of infections, from meningitis to typhoid fever to conjunctivitis. Unfortunately, serious side effects, such as lethal grey baby syndrome, and suppression of bone marrow production, have limited its clinical role. Chloramphenicol also causes anaemia in two different ways. One mechanism involves the targeting of mitochondrial ribosomes within hematopoietic stem cells, causing a reversible, dose-dependent suppression of blood cell production. Once chloramphenicol dosing is discontinued, blood cell production returns to normal. This mechanism highlights the similarity between 70S ribosomes of bacteria and the 70S ribosomes within our mitochondria. The second mechanism of anaemia is idiosyncratic (i.e., the mechanism is not understood), and involves an irreversible lethal loss of blood cell production known as aplastic anaemia. This mechanism of aplastic anaemia is not dose dependent and can develop after therapy has stopped. Because of toxicity concerns, chloramphenicol usage in humans is now rare in the United States and is limited to severe infections unable to be treated by less toxic antibiotics. Because its side effects are much less severe in animals, it is used in veterinary medicine.
The oxazolidinones, including linezolid, are a new broad-spectrum class of synthetic protein synthesis inhibitors that bind to the 50S ribosomal subunit of both gram-positive and gram-negative bacteria. However, their mechanism of action seems somewhat different from that of the other 50S subunit-binding protein synthesis inhibitors already discussed. Instead, they seem to interfere with formation of the initiation complex (association of the 50S subunit, 30S subunit, and other factors) for translation, and they prevent translocation of the growing protein from the ribosomal A site to the P site. Table 15.3 summarizes the protein synthesis inhibitors.
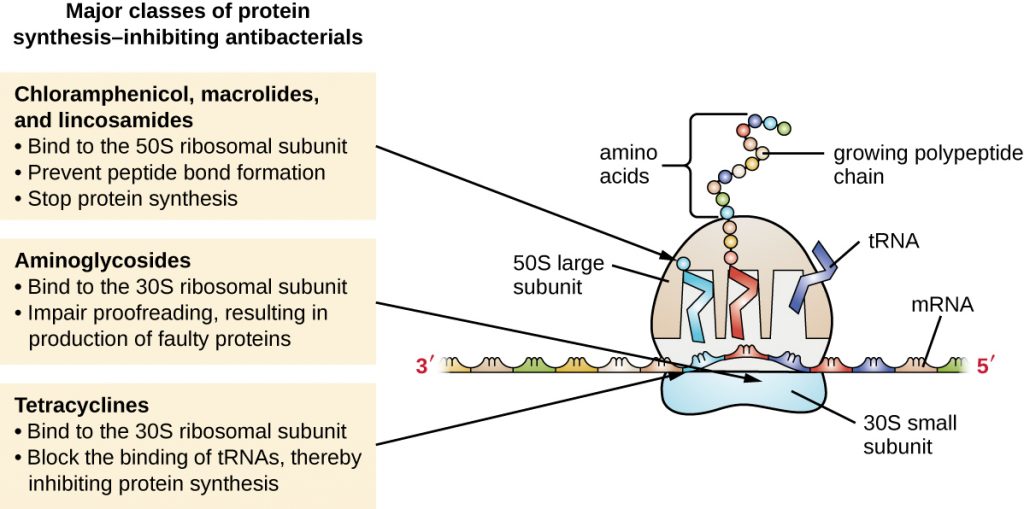
Table 15.3. Drugs that Inhibit Bacterial Protein Synthesis
| Drugs That Inhibit Bacterial Protein Synthesis | |||||
|---|---|---|---|---|---|
| Molecular Target | Mechanism of Action | Drug Class | Specific Drugs | Bacteriostatic or Bactericidal | Spectrum of Activity |
| 30S subunit | Causes mismatches between codons and anticodons, leading to faulty proteins that insert into and disrupt cytoplasmic membrane | Aminoglycosides | Streptomycin, gentamicin, neomycin, kanamycin | Bactericidal | Broad spectrum |
| Blocks association of tRNAs with ribosome | Tetracyclines | Tetracycline, doxycycline, tigecycline | Bacteriostatic | Broad spectrum | |
| 50S subunit | Blocks peptide bond formation between amino acids | Macrolides | Erythromycin, azithromycin, telithromycin | Bacteriostatic | Broad spectrum |
| Lincosamides | Lincomycin, clindamycin | Bacteriostatic | Narrow spectrum | ||
| Not applicable | Chloramphenicol | Bacteriostatic | Broad spectrum | ||
| Interferes with the formation of the initiation complex between 50S and 30S subunits and other factors. | Oxazolidinones | Linezolid | Bacteriostatic | Broad spectrum | |
- Compare and contrast the different types of protein synthesis inhibitors.
Inhibitors of Membrane Function
A small group of antibacterials target the bacterial membrane as their mode of action (Table 15.4). The polymyxins are natural polypeptide antibiotics that were first discovered in 1947 as products of Bacillus polymyxa; only polymyxin B and polymyxin E (colistin) have been used clinically. They are lipophilic with detergent-like properties and interact with the lipopolysaccharide component of the outer membrane of gram-negative bacteria, ultimately disrupting both their outer and inner membranes and killing the bacterial cells. Unfortunately, the membrane-targeting mechanism is not a selective toxicity, and these drugs also target and damage the membrane of cells in the kidney and nervous system when administered systemically. Because of these serious side effects and their poor absorption from the digestive tract, polymyxin B is used in over-the-counter topical antibiotic ointments (e.g., Neosporin), and oral colistin was historically used only for bowel decontamination to prevent infections originating from bowel microbes in immunocompromised patients or for those undergoing certain abdominal surgeries. However, the emergence and spread of multidrug-resistant pathogens has led to increased use of intravenous colistin in hospitals, often as a drug of last resort to treat serious infections. The antibacterial daptomycin is a cyclic lipopeptide produced by Streptomyces roseosporus that seems to work like the polymyxins, inserting in the bacterial cell membrane and disrupting it. However, in contrast to polymyxin B and colistin, which target only gram-negative bacteria, daptomycin specifically targets gram-positive bacteria. It is typically administered intravenously and seems to be well tolerated, showing reversible toxicity in skeletal muscles.
Table 15.4. Drugs that Inhibit Bacterial Membrane Function
| Drugs That Inhibit Bacterial Membrane Function | ||||
|---|---|---|---|---|
| Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity | Clinical Use |
| Interacts with lipopolysaccharide in the outer membrane of gram-negative bacteria, killing the cell through the eventual disruption of the outer membrane and cytoplasmic membrane | Polymyxins | Polymyxin B | Narrow spectrum against gram-negative bacteria, including multidrug-resistant strains | Topical preparations to prevent infections in wounds |
| Polymyxin E (colistin) | Narrow spectrum against gram-negative bacteria, including multidrug-resistant strains | Oral dosing to decontaminate bowels to prevent infections in immunocompromised patients or patients undergoing invasive surgery/procedures. | ||
| Intravenous dosing to treat serious systemic infections caused by multidrug-resistant pathogens | ||||
| Inserts into the cytoplasmic membrane of gram-positive bacteria, disrupting the membrane and killing the cell | Lipopeptide | Daptomycin | Narrow spectrum against gram-positive bacteria, including multidrug-resistant strains | Complicated skin and skin-structure infections and bacteraemia caused by gram-positive pathogens, including MRSA |
- How do polymyxins inhibit membrane function?
Inhibitors of Nucleic Acid Synthesis
Some antibacterial drugs work by inhibiting nucleic acid synthesis (Table 15.5). For example, metronidazole is a semisynthetic member of the nitroimidazole family that is also an antiprotozoan. It interferes with DNA replication in target cells. The drug rifampin is a semisynthetic member of the rifamycin family and functions by blocking RNA polymerase activity in bacteria. The RNA polymerase enzymes in bacteria are structurally different from those in eukaryotes, providing for selective toxicity against bacterial cells. It is used for the treatment of a variety of infections, but its primary use, often in a cocktail with other antibacterial drugs, is against mycobacteria that cause tuberculosis. Despite the selectivity of its mechanism, rifampin can induce liver enzymes to increase metabolism of other drugs being administered (antagonism), leading to hepatotoxicity (liver toxicity) and negatively influencing the bioavailability and therapeutic effect of the companion drugs.
One member of the quinolone family, a group of synthetic antimicrobials, is nalidixic acid. It was discovered in 1962 as a byproduct during the synthesis of chloroquine, an antimalarial drug. Nalidixic acid selectively inhibits the activity of bacterial DNA gyrase, blocking DNA replication. Chemical modifications to the original quinolone backbone have resulted in the production of fluoroquinolones, like ciprofloxacin and levofloxacin, which also inhibit the activity of DNA gyrase. Ciprofloxacin and levofloxacin are effective against a broad spectrum of gram-positive or gram-negative bacteria, and are among the most commonly prescribed antibiotics used to treat a wide range of infections, including urinary tract infections, respiratory infections, abdominal infections, and skin infections. However, despite their selective toxicity against DNA gyrase, side effects associated with different fluoroquinolones include phototoxicity, neurotoxicity, cardiotoxicity, glucose metabolism dysfunction, and increased risk for tendon rupture.
Table 15.5. Drugs that Inhibit Bacterial Nucleic Acid Synthesis
| Drugs That Inhibit Bacterial Nucleic Acid Synthesis | ||||
|---|---|---|---|---|
| Mechanisms of Action | Drug Class | Specific Drugs | Spectrum of activity | Clinical Use |
| Inhibits bacterial RNA polymerase activity and blocks transcription, killing the cell | Rifamycin | Rifampin | Narrow spectrum with activity against gram-positive and limited numbers of gram-negative bacteria. Also active against Mycobacterium tuberculosis. | Combination therapy for treatment of tuberculosis |
| Inhibits the activity of DNA gyrase and blocks DNA replication, killing the cell | Fluoroquinolones | Ciprofloxacin, ofloxacin, moxifloxacin | Broad spectrum against gram-positive and gram-negative bacteria | Wide variety of skin and systemic infections |
- Why do inhibitors of bacterial nucleic acid synthesis not target host cells?
Inhibitors of Metabolic Pathways
Some synthetic drugs control bacterial infections by functioning as antimetabolites, competitive inhibitors for bacterial metabolic enzymes (Table 15.6). The sulfonamides (sulfa drugs) are the oldest synthetic antibacterial agents and are structural analogues of para-aminobenzoic acid (PABA), an early intermediate in folic acid synthesis (Figure 15.12). By inhibiting the enzyme involved in the production of dihydrofolic acid, sulfonamides block bacterial biosynthesis of folic acid and, subsequently, pyrimidines and purines required for nucleic acid synthesis. This mechanism of action provides bacteriostatic inhibition of growth against a wide spectrum of gram-positive and gram-negative pathogens. Because humans obtain folic acid from food instead of synthesizing it intracellularly, sulfonamides are selectively toxic for bacteria. However, allergic reactions to sulfa drugs are common. The sulfones are structurally similar to sulfonamides but are not commonly used today except for the treatment of Hansen’s disease (leprosy).
Trimethoprim is a synthetic antimicrobial compound that serves as an antimetabolite within the same folic acid synthesis pathway as sulfonamides. However, trimethoprim is a structural analogue of dihydrofolic acid and inhibits a later step in the metabolic pathway (Figure 15.12). Trimethoprim is used in combination with the sulfa drug sulfamethoxazole to treat urinary tract infections, ear infections, and bronchitis. As discussed, the combination of trimethoprim and sulfamethoxazole is an example of antibacterial synergy. When used alone, each antimetabolite only decreases production of folic acid to a level where bacteriostatic inhibition of growth occurs. However, when used in combination, inhibition of both steps in the metabolic pathway decreases folic acid synthesis to a level that is lethal to the bacterial cell. Because of the importance of folic acid during fetal development, sulfa drugs and trimethoprim use should be carefully considered during early pregnancy.
The drug isoniazid is an antimetabolite with specific toxicity for mycobacteria and has long been used in combination with rifampin or streptomycin in the treatment of tuberculosis. It is administered as a prodrug, requiring activation through the action of an intracellular bacterial peroxidase enzyme, forming isoniazid-nicotinamide adenine dinucleotide (NAD) and isoniazid-nicotinamide adenine dinucleotide phosphate (NADP), ultimately preventing the synthesis of mycolic acid, which is essential for mycobacterial cell walls. Possible side effects of isoniazid use include hepatotoxicity, neurotoxicity, and hematologic toxicity (anaemia).
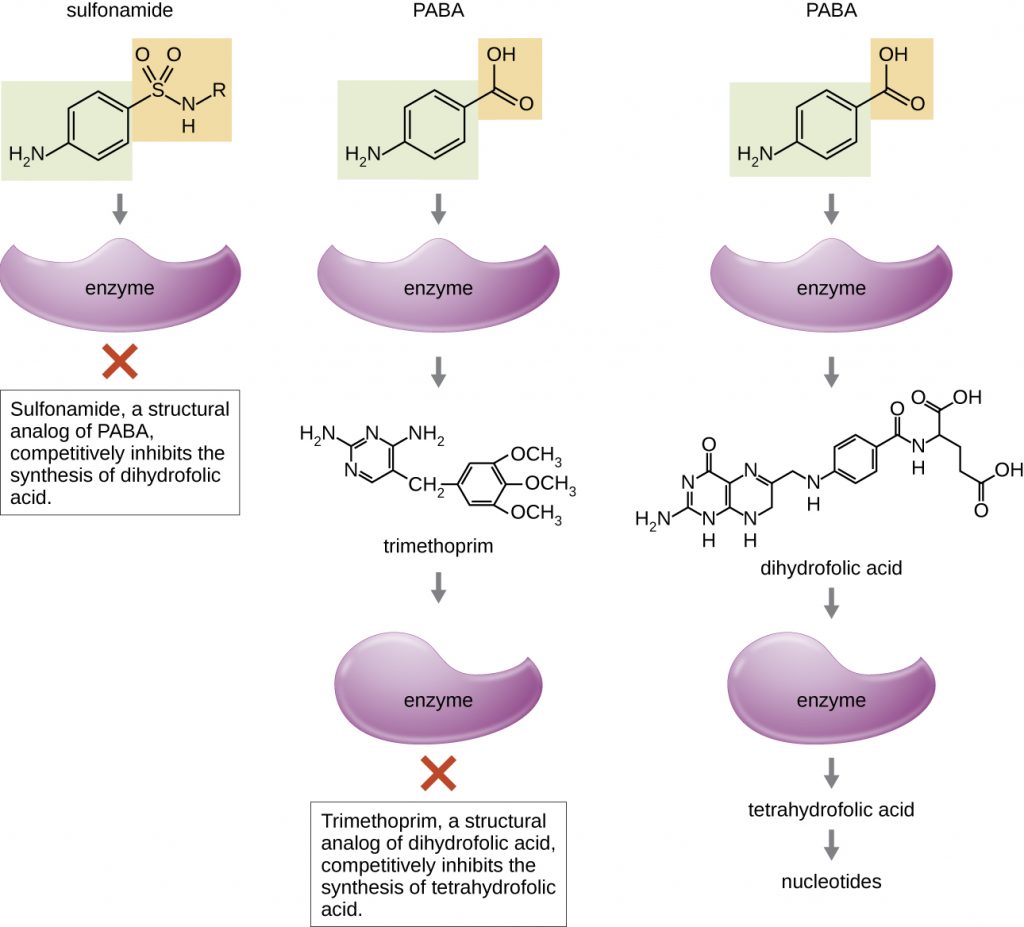
Table 15.6. Antimetabolite Drugs
| Antimetabolite Drugs | ||||
|---|---|---|---|---|
| Metabolic Pathway Target | Mechanism of Action | Drug Class | Specific Drugs | Spectrum of Activity |
| Folic acid synthesis | Inhibits the enzyme involved in production of dihydrofolic acid | Sulfonamides | Sulfamethoxazole | Broad spectrum against gram-positive and gram-negative bacteria |
| Sulfones | Dapsone | |||
| Inhibits the enzyme involved in the production of tetrahydrofolic acid | Not applicable | Trimethoprim | Broad spectrum against gram-positive and gram-negative bacteria | |
| Mycolic acid synthesis | Interferes with the synthesis of mycolic acid | Not applicable | Isoniazid | Narrow spectrum against Mycobacterium spp., including M. tuberculosis |
- How do sulfonamides and trimethoprim selectively target bacteria?
Inhibitor of ATP Synthase
Bedaquiline, representing the synthetic antibacterial class of compounds called the diarylquinolones, uses a novel mode of action that specifically inhibits mycobacterial growth. Although the specific mechanism has yet to be elucidated, this compound appears to interfere with the function of ATP synthases, perhaps by interfering with the use of the hydrogen ion gradient for ATP synthesis by oxidative phosphorylation, leading to reduced ATP production. Due to its side effects, including hepatotoxicity and potentially lethal heart arrhythmia, its use is reserved for serious, otherwise untreatable cases of tuberculosis.

To learn more about the general principles of antimicrobial therapy and bacterial modes of action, visit Michigan State University’s Antimicrobial Resistance Learning Site, particularly pages 6 through 9.
CLINICAL FOCUS: Part 2
Reading thorough Marisa’s health history, the doctor noticed that during her hospitalization in Vietnam, she was catheterized and received the antimicrobial drugs ceftazidime and metronidazole. Upon learning this, the doctor ordered a CT scan of Marisa’s abdomen to rule out appendicitis; the doctor also requested blood work to see if she had an elevated white blood cell count, and ordered a urine analysis test and urine culture to look for the presence of white blood cells, red blood cells, and bacteria.
Marisa’s urine sample came back positive for the presence of bacteria, indicating a urinary tract infection (UTI). The doctor prescribed ciprofloxacin. In the meantime, her urine was cultured to grow the bacterium for further testing.
- What types of antimicrobials are typically prescribed for UTIs?
- Based upon the antimicrobial drugs she was given in Vietnam, which of the antimicrobials for treatment of a UTI would you predict to be ineffective?
Jump to the next Clinical Focus box. Go back to the previous Clinical Focus box.
Key Takeaways
- Antibacterial compounds exhibit selective toxicity, largely due to differences between prokaryotic and eukaryotic cell structure.
- Cell wall synthesis inhibitors, including the β-lactams, the glycopeptides, and bacitracin, interfere with peptidoglycan synthesis, making bacterial cells more prone to osmotic lysis.
- There are a variety of broad-spectrum, bacterial protein synthesis inhibitors that selectively target the prokaryotic 70S ribosome, including those that bind to the 30S subunit (aminoglycosides and tetracyclines) and others that bind to the 50S subunit (macrolides, lincosamides, chloramphenicol, and oxazolidinones).
- Polymyxins are lipophilic polypeptide antibiotics that target the lipopolysaccharide component of gram-negative bacteria and ultimately disrupt the integrity of the outer and inner membranes of these bacteria.
- The nucleic acid synthesis inhibitors rifamycins and fluoroquinolones target bacterial RNA transcription and DNA replication, respectively.
- Some antibacterial drugs are antimetabolites, acting as competitive inhibitors for bacterial metabolic enzymes. Sulfonamides and trimethoprim are antimetabolites that interfere with bacterial folic acid synthesis. Isoniazid is an antimetabolite that interferes with mycolic acid synthesis in mycobacteria.
Multiple Choice
Fill in the Blank
True/False
Short Answer
- If human cells and bacterial cells perform transcription, how are the rifamycins specific for bacterial infections?
- What bacterial structural target would make an antibacterial drug selective for gram-negative bacteria? Provide one example of an antimicrobial compound that targets this structure.
Critical Thinking
- In considering the cell structure of prokaryotes compared with that of eukaryotes, propose one possible reason for side effects in humans due to treatment of bacterial infections with protein synthesis inhibitors.
Media Attributions
- OSC_Microbio_14_02_Modes
- OSC_Microbio_14_02_BetaLactam
- OSC_Microbio_14_02_ProtSynInh
- OSC_Microbio_14_02_SulfTri
- microbiology sign © Nick Youngson
