17 Organic Reactions
17.1: Reactions
Chemical reactions in organic compounds tend to occur at reactive sites on the molecules, where there is buildup of either positive or negative charge. The charges in the substrate (the main organic molecule being reacted) will occur depending on the structure composition of the molecule. For example, a carbon-carbon double bond has two pairs of electrons in close proximity to each other, making for a concentration of negative charge whereas, if there is an electronegative atom bonded to a carbon, it will tend to inductively withdraw some of the electron density away from the carbon creating a positive charge concentration on that carbon.
17.1.1 Curly arrows
When we draw out the reaction mechanisms, it’s important to keep track of where the electrons are, especially the ones involved in forming and breaking bonds. Generally, we can write an overall process as a simple balanced chemical reaction, but that reaction often does not happen in a single step as is implied by the single equation. Many reactions take several steps to occur and each step may involve the creation or destruction of bonds. To keep track of these changes, we use curly arrows to represent the movement of electrons. Remember that the electrons are thousands of times lighter than the nuclei of the atoms themselves so although we often think and speak as if it’s the atoms that do the moving, we are better off to imagine the electrons as doing the moving while the atoms are relatively motionless. We have explored this concept earlier when we looked at resonance structures of a single molecule or polyatomic ion species. The resonance structures involved electrons jumping around at thousands of times the rate that the atoms themselves could move, so it was just simpler to assume the atoms don’t move. We will be in essence extending this concept here but now, the electrons will be moving in such a way as to create or destroy bonds.
It’s important to remember that every curly arrow must start at a pair of electrons, either in a bond, or a lone pair on an atom. The arrow must end in such a way that the receiving location can accommodate the electrons without violating any rules. If the electron pair originated as a bond, then it will end on an atom, which will then develop a negative charge relative to its original charge and the bond will be broken.
Curly arrows can also be used to show bonds forming when electrons move into the space between two atoms.
Sometimes, the curly arrows can represent a bond breaking and a new bond forming in a single step.![]()
Sometimes, bonds are broken and formed homolytically, meaning equally between the two atoms. In this case, the pair of electrons does not stay together through the process. If a bond is broken via this kind of process the resulting new species will have unpaired electrons on the two atoms that were bonded. They may not have changed their charge but they will both be radicals, and generally very reactive. The reverse of this is the formation of a bond by two radicals coming together and the individual electrons on the radicals jump into the same space to make a bond. Alternatively, a pair of electrons on one molecule might be split up so that the radical joins with one of them and a new radical is formed. This kind of propagation of the radical is found in many kinds of polymerization reactions. The curly arrows that are used in this case is a harpoon, with the arrow head only on one side to indicate only one electron moved.
In the cracking reaction, the long-chain hydrocarbon is heated until a C-C bond breaks. Since the two bonded carbons are equal, there is a chance that the bond will break homolytically to make two radicals. You can see both homolytic cleavage and bond creation in the cracking mechanism.
In the polymerization of the ethene molecule (also called ethylene) to make polyethylene, the first step is to break the π-bond homolytically with the help of a catalyst, like a metal-chloride complex, to make ethylene monomer with two end carbons as radicals. This would normally just recombine rapidly to reform the bond but if the pressure is high enough, a second ethylene monomer might bump into it before it can recombine. The ethylene radical then induces other ethylene monomers it comes in contact with to cleave its π-bond spontaneously and form a new bond joining the two monomers into a longer chain (polymer). This repeats indefinitely until something else reacts with the radical ends of the chain to terminate the polymerization. This could be something as simple as chlorine itself.
17.1.2 How to identify reactive sites
Generally, in organic chemistry reactions, good sites for an electron pair to jump to will be slightly positive. This is most often found in Carbon chains when the carbon is bonded to a heteroatom that withdraws electron density off the carbon. Thus, a more electronegative atom like N, O or F, can induce a positive charge on the α-carbon (the one bonded to the heteroatom) and this site will be the electrophile that accepts the electron from the nucleophile.
Consider an organic molecule with several functional groups. The first job is to identify all the functional groups and then to figure out how those groups might induce reactivity.
Remember:
Heteroatoms like N, O, S, etc. can have lone pairs that can be donated (Nucleophile or Lewis base).
Double or triple bonds involve a high concentration of electrons and the π-bonds can act as a nucleophile.
Electronegative atoms like N, O, F, etc. can induce positive charges on the neighbors as they pull electron density towards themselves. you can use these identifiers to see where electron pairs might originate (nucleophiles) and where they might land (electrophiles).
Take a moment and look at this molecule to identify all the functional groups and see if you can figure out where the reactivity might lie.

This next image highlights the possible reactive sites based on the functional groups that have been identified. We can see three places from which an electron pair might be donated in a reaction (the lone pairs on Cl and on N and the π-bond) and a place where electrons might land (the C attached to the Cl, which is positively charged).

Finally, when we draw the actual reaction of molecules, we sometimes get a bit lazy and don’t draw everything. In the molecule above, for example, the hydrogens (and carbons) are not drawn but you can figure out where they are. There is a carbon at every joint between the line segments and each carbon needs to have 4 bonds, if it’s neutral.
Here is an example of a reaction showing a Lewis base forming a bond with a proton, which breaks its bond to the oxygen. The resulting BH+ species is often not even shown. Lone pairs that are not involved in the reaction are often not drawn.
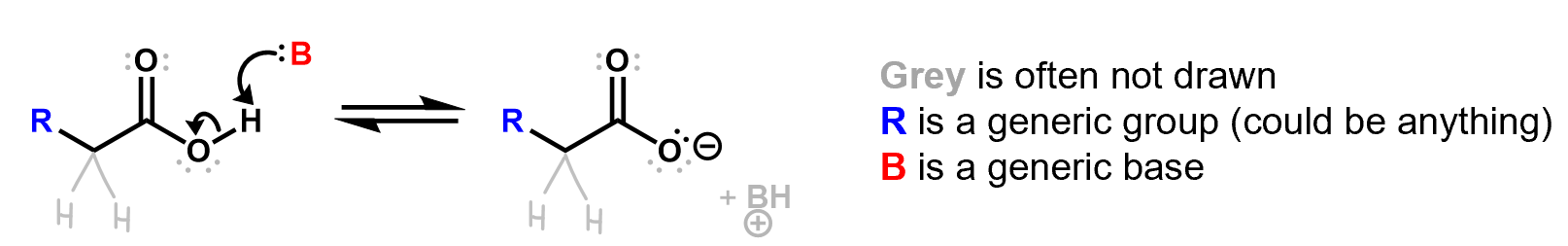
Be careful when you’re reading reaction mechanisms to fill in all these details (at least in your mind) before you get confused. Experienced chemists can do this automatically without much thought but you may need to actively work on it at first.
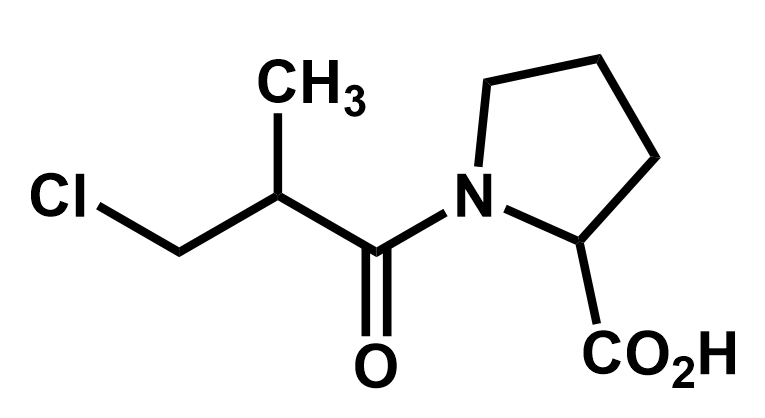
Practice 1: Identify the functional groups on this molecule and mark all the places that may develop charges, in anticipation of drawing curly arrows.
17.1.3 Nucleophiles and Electrophiles
The reaction is often initiated by oppositely charged molecules or atoms approaching these charged entities. A molecule or portion of a molecule that has excess negative charge is attracted to the positive charges (the nuclei) of other molecules. They are called nucleophiles. A nucleophile [ nucleus lover ] can be any neutral or charged species that has a concentration of electron density and also has a lone pair it can use to attack the positive part of the other molecule (Because it’s attracted to it). The positive fragment of the molecule is called an electrophile [ electron lover ]. It attracts any concentration of negative charge (nucleophiles).
Some nucleophiles are better than others but all have one thing in common. They are essentially Lewis Bases. There are several points you can look for, to rank a list of nucleophiles.
- Smaller molecules tend to be better nucleophiles. This is mostly because they often have to fit into small spaces to get close to the nucleus they are reacting with. Look for smaller molecules and ions with lone pairs to make good nucleophiles.
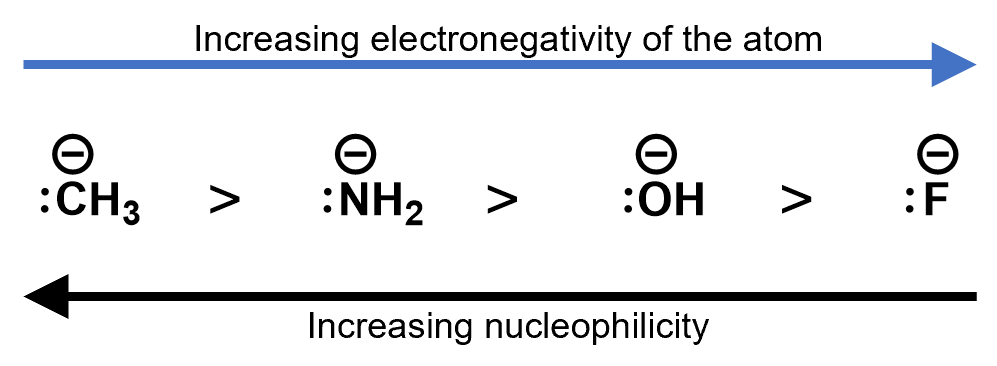
- Electronegativity. This may seem counter intuitive but if an atom is too electronegative, it will tend to hold onto its electron pairs and not react as a nucleophile as well as a similar atom that is less electronegative. But the atoms that do act as nucleophiles tend to be some of the more electronegative atoms. Thus, for example, as you go from C > N > O > F in the periodic table, the nucleophilicity of the atoms drops.
- Polarizability. Atoms that are more polarizable (meaning the electron cloud can move about easier on it, the better it is as a nucleophile. Generally, larger atoms are more polarizable because the valence electrons are further from the nucleus. Thus, looking at the halogens, they rank from best to worst as I– > Br– > Cl– > F–. Note that this seems to contrast with point 1. The key is 1 is talking about having enough room for the nucleophile to get into the reactive site on the carbon backbone while 3 is talking about the ability of the electrons to move around on an atom within that nucleophilic species. So, while I– is bigger than Cl– , the ability of the electrons to move around (and reach out a bit on the I–) overrides steric issues. These do indeed compete and sometimes, It’s not necessarily clear cut which is the better. In this course, we’ll keep to simple options where the answer is more clear.
- Solvent effects. Some nucleophiles will interact with the solvent and not work well as a nucleophile. So the particular strength of the nucleophile will depend on the solvent. But that’s for a later class.
Notice that ions like I–, Br–, and Cl– are conjugates of strong acids, meaning they are very poor bases but they are excellent nucleophiles. The weaker the base, the stronger the nucleophile. Generally, a nucleophile is also a base but strong bases make poorer nucleophiles and vice versa. We tend to call it a nucleophile when it reacts with a carbon (called the α-carbon) on a substrate molecule in a substitution reaction (See SN1 and SN2 below). We call it a base if it reacts with a H+ on the β-carbon in an elimination reaction (see E1 and E2 below). The reaction (a pair of electrons jumping onto an orbital on a different atom to make a bond) is the same either way. The nucleophile or Lewis base donates a pair of electrons to a carbon (Substitution) or a hydrogen (Elimination) on the substrate (Lewis acid) to create a new bond.
The following table contains a list of different nucleophiles and some approximate ranking of their strengths
| Very good nucleophiles | HS–, I–, RS.– |
| Good nucleophiles | Br–, OH–, RO–, CN–, N3– |
| nucleophiles | NH3, Cl–, F–, RCOO– |
| weak nucleophiles | H2O, ROH |
| very weak nucleophiles | RCOOH |
Notice that not all nucleophiles are negatively charged but all of them have at least one lone pair so they can act as an electron pair donor (Lewis base or nucleophile). In addition to these listed here, are any molecule that has double or triple bonds or lone pairs with high concentration of electrons.
Electrophiles, the entities nucleophiles react with, tend to be electron deficient in some way. They have partially or fully positive charges. They accept the electrons from the nucleophiles. These can include atoms that are bonded to more electronegative atoms or other kinds of electron withdrawing groups.
Electrophiles can be positively charged, like
H+, NO+, R+, protonated amines, like R-NH2+, and carbo-cations like CH3C(+)=O
They can be electrically neutral but have empty orbitals, like
Lewis acids: BF3, AlCl3, SO3, ZnCl2, BeCl2, FeCl3.
Carbon atoms attached to a more electronegative atom can act as electrophiles because they will have a partially positive charge due to induction.
Look for electron pair motion in some of these reactions to see which are the nucleophiles and which are the electrophiles.
17.2: Reactions of Alkanes
The most easily recognized reaction of an alkane is when it is burned in air to give carbon dioxide and water. Actually, this is far from a single step reaction, even though it is often depicted as such. This is the ultimate in a series of oxidation reactions of hydrocarbons. Lesser oxidation reactions would form alcohols, aldehydes, ketones and acids. We won’t delve into those in this course. Here is a simple reaction showing the combustion of propane. It hides a much more complex multi-step path that can end, with insufficient oxygen, heat or time, in many different products along the path to full oxidation products carbon dioxide and water, shown here.

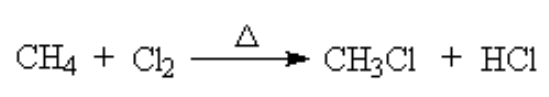
Alkanes can react with halogen at high T. This is called a halogenation reaction. Here, we see the chlorination of methane. (See details here)
Cracking (in oil refining, used to reduce the molar mass of the compounds so they can be used as gasoline, etc.) Cracking Explained Details
dehydrogenation (removal of a hydrogen molecule H2)
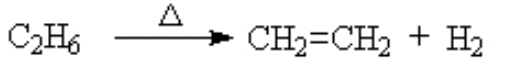
17.3: Reactions of alkenes and alkynes
Double and triple bonds are good nucleophiles. They are a concentration of electron density (more than one electron pair in the one domain) and in particular, the π-electrons are less strongly held and can be affected.
17.3.1: Addition reactions
Addition reactions all have a similar form with each other. The pair of electrons in the π-bond latches onto one atom in the molecule, breaking the bond to the second atom in the pair. That second atom, is electron rich and attacks the carbo-cation formed by the initial attack of the π electron pair.
Bromine (any halogen molecule) can add symmetrically across a double (or a triple) bond. Here is the bromination of ethene.
The hydrogenation reaction will not occur without a catalyst. If you heat an alkane, the spontaneous reaction is the reverse, dehydrogenation. With a catalyst, we can force the reaction in this direction. (see details)
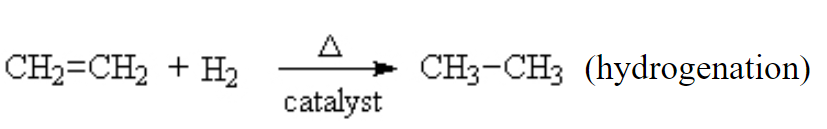
Markovnikov addition. Finally, we come to one of the more interesting of addition reactions. We briefly saw it above when we added HCl to ethene. But what if the alkene is not symmetrical? in this case, there is a general rule, which informs us as to the most likely outcome. It’s called Markovnikov’s rule. It was noted that when making this addition, the products were not equally divided between the two possibilities. The proton from the HCl tended to go to the Carbon on the ene that already had the larger number of protons. Thus, in this example, The =CH2 group got the hydrogen to become a –CH3. There is always a smaller amount of the product where the chlorine ended up on that side but the dominant product was always the same.
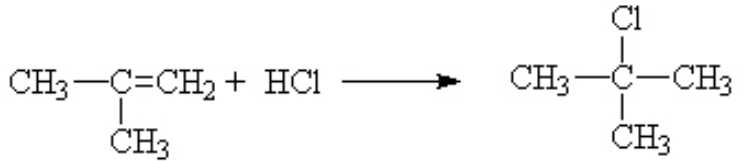
That is, it is the same as long as the substituent on the other side of the ene is an electron donating group like methyl. If you had substituted an electron withdrawing group, like a halogen, then the opposite result was dominant. The proton went mostly to the side with the lesser number protons already.
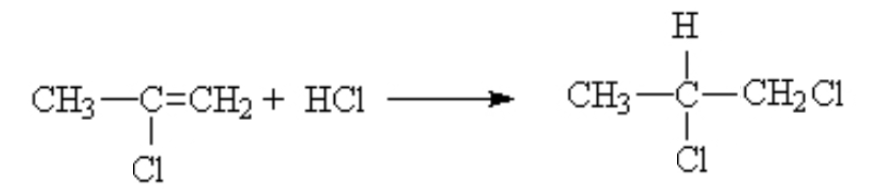
This was deemed anti-Markovnikov behaviour. Later, when the mechanism was worked out, the reason for this behaviour as better understood.
Click here to See the details for more explanation on the Markovnikov addition.
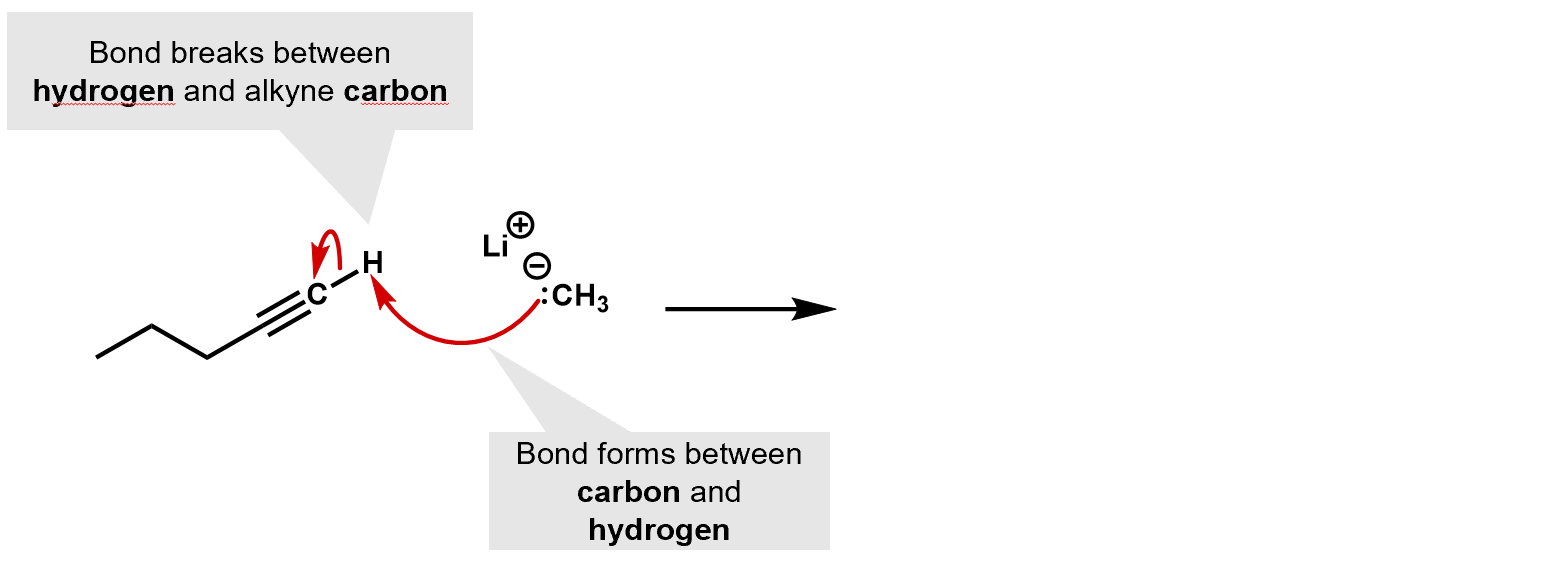
Alkynes can react with the strong base sodium amide, to produce the alkylide ion (in this case, the acetylene produces the acetylide ion, a base that is far stronger than even NaOH) and as a group. Because they are such a strong base, they tend to be very selective nucleophiles.
![]()
17.3.2: Substitution Reactions
In a substitution reaction one substituent is substituted for another. A very common type of substitution involves the replacement of a leaving group X (X is something that can leave the molecule relatively easily, such as a halide) by a nucleophile Nu (the suffix ~’phile’ implies “attraction to” so a nucleophile is attracted to the positive charge of a nucleus). I will use Nu to stand for the generic nucleophile. Normally, a nucleophile is negatively charged or polar with lone pairs of electrons. These can be used to form new a new bond with the nucleus (carbon atom) involved. There are two types of Nucleophilic Substitution, SN1 and SN2:
17.3.2.1 SN1 Reaction mechanism
In an SN1 process, there are two steps involved. The first step (the slow step) involves only the starting molecule, the nucleophile is not involved. Hence the 1 of SN1 means The reaction is first order (See Kinetics Chapter). It also means that 1 molecule is involved (unimolecular) in the slow, rate-determining step of the mechanism. In this step, the leaving group takes its pair of electrons and leaves behind a positively charged trigonal-planar α-carbon (alias carbocation, also alias carbonium ion)[1]. Then the nucleophile bonds to the carbocation. In this case, because the intermediate is symmetrical (trigonal planar) before the nucleophilic attack, there are two possibly products, depending on the side from which the nucleophile attacks.
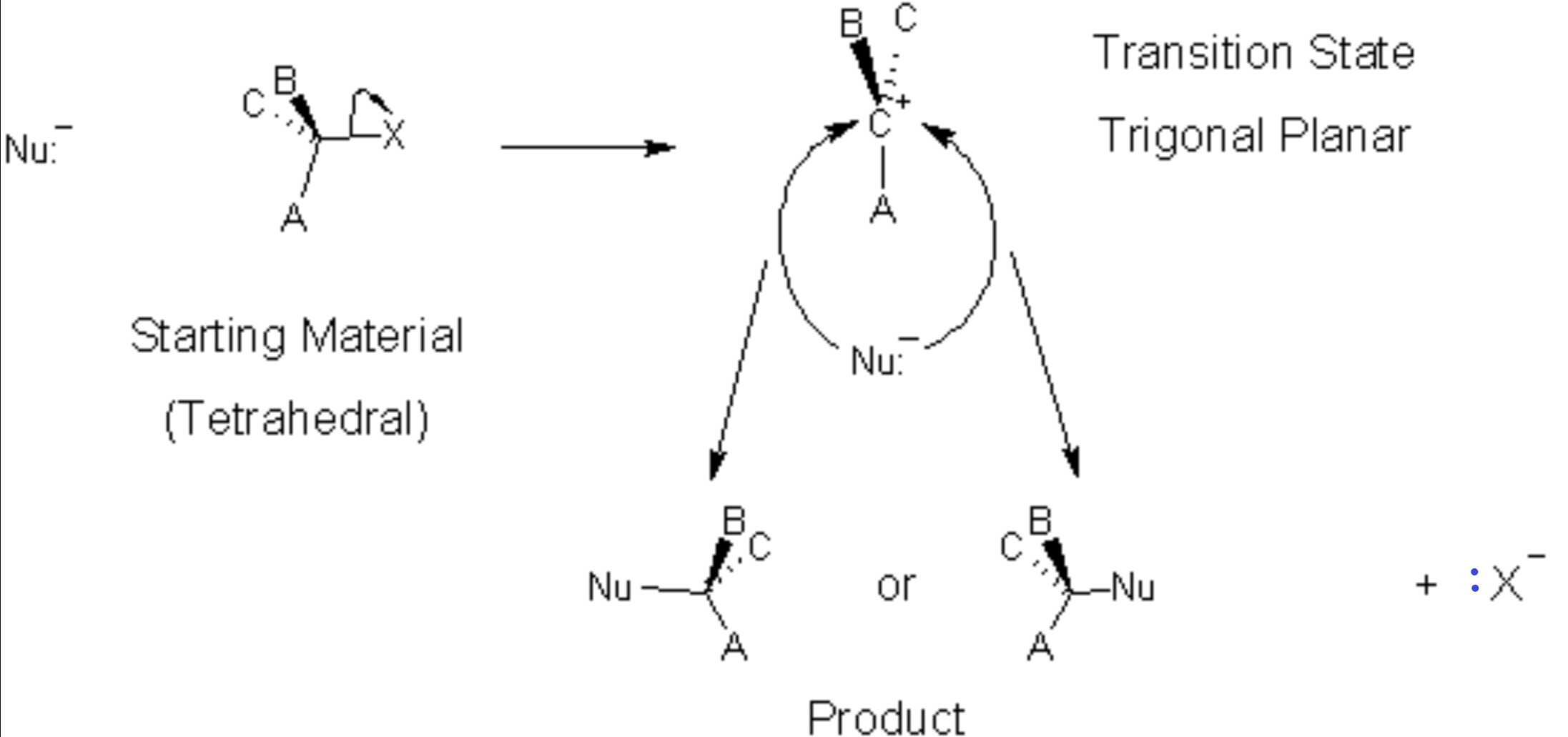
In this reaction mechanism, because the nucleophile is just as likely to attack from the left as from the right, we will get a 50:50 mixture of the two products. If the reactant was chiral (optically active) then the two products are both chiral but of opposite chirality. The mixture will display no optical activity since the two products’ optical activities cancel each other out. This is a racemic mixture.
So, we need to consider the reactivity of the nucleophile/base but also, the structure of the substrate is important. The carbocation is most stable if it is highly substituted (methyl groups are electron donating) so a tertiary carbon is more likely to undergo SN1 reaction than a primary carbon, which has no electron donor groups to stabilize the carbocation. In fact, the primary carbocation is so unstable as to make an SN1 reaction very unlikely. Thus, as far as the substrate goes, the relative reactivity is as follows:
(CH3)3C—LG > (CH3)2CH—LG > CH3CH2—LG > CH3—LG.
Where LG is the leaving group. The α-carbon is boldfaced.
17.3.2.2 SN2 Reaction Mechanism
In the SN2 process, the nucleophile bonds to the carbon while the leaving group breaks its bond in a concerted step. This first step (slow, rate-determining step) involves 2 molecules simultaneously (bimolecular) so the reaction is second order (see Kinetics chapter). The starting material and the nucleophile. The reaction is second order. Hence the 2 in SN2.
At the intermediate (or transition state), the positions of constituents ABC move to a planar orientation with the Nu on one side and the leaving group X on the other to form a trigonal bipyramidal intermediate (transition state). As the nucleophile moves closer with its lone pair, the leaving group departs, taking its bonding pair as a lone pair and taking the excess charge in the process. The spatial orientation of the groups ABC is reversed in the process. In the case of an optical isomer (chiral molecule) the chirality (direction that light is rotated) can be reversed.
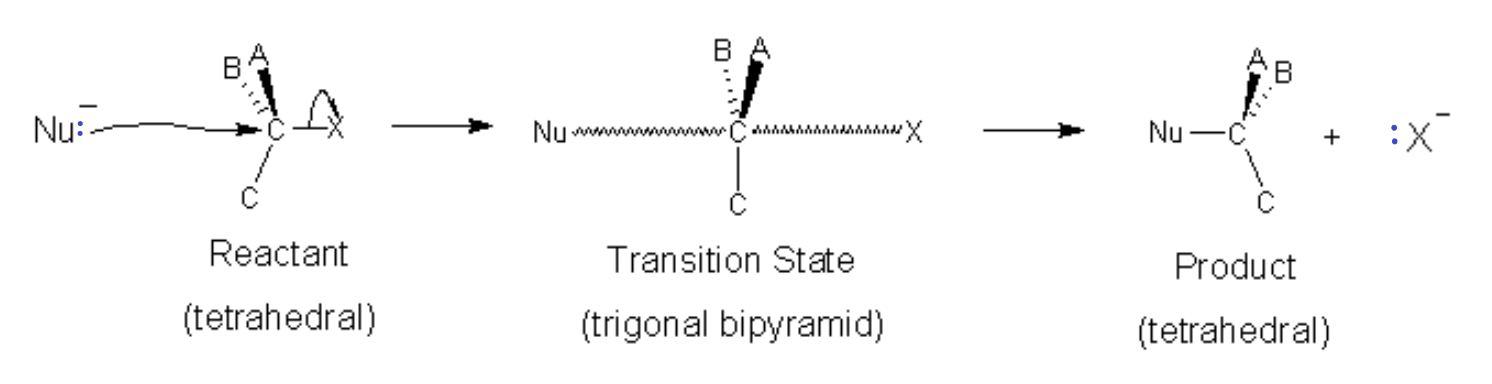
Note that in the case of SN2 reactions, the α-carbon is tetrahedral when the Nucleophilic attack occurs. This means that there is greater possibility of steric hindrance. If the carbon on which the reaction should occur is tertiary, meaning substituents A,B,C are all alkyl groups, then there will be little room for the nucleophile to get near the Carbon substrate and an SN2 reaction is unlikely to happen. An SN1 reaction might be possible though because the leaving group will allow the tetrahedral carbon to open up into a trigonal planar carbocation.
Notice that in both of these reactions, the leaving group contains a lone pair and a negative charge. It is also a nucleophile, albeit, a weaker nucleophile than the one that replaced it. The leaving group can be ranked according to how good a leaving group they are just like the ranking of the nucleophilic strength, except the order would be reversed. A strong nucleophile would not make a good leaving group but a weak nucleophile will make an excellent leaving group. That makes sense because the strong nucleophile is less stable. It will react readily with the substrate and is therefore more likely to attach itself to the substrate whereas a weak nucleophile is more content to be kicked out and remain in solution because it is more stable.
When we consider the substrate in an SN2 reaction, steric hindrance on the α-carbon becomes more important. If groups A,B,C are bulky then there will be no room for the nucleophile to get close to the α-carbon. Thus, for SN2 reactions, the order of reactivity of a carbon as a function of how much substitution is the reverse of what you saw for SN1:
(CH3)3C—LG < (CH3)2CH—LG < CH3CH2—LG < CH3—LG.
This reversal of order is so important that we can generalize and say that no matter the nucleophile and leaving group, a tertiary carbon will only undergo SN1 but a primary carbon will only undergo SN2. A secondary carbon might go either way, depending on the nucleophile and leaving group strengths.
17.3.3: Elimination Reaction (opposite of addition)
A saturated hydrocarbon (as many hydrogens as possible) is converted to an unsaturated hydrocarbon by ‘eliminating’ a hydrogen molecule. This is also called dehydrogenation. This kind of reaction involves lots of heat and is generally not well controlled, in longer chain alkanes, the particular place where the hydrogen is removed is quite random. The dehydrogenation of a long-chain alkane in a fatty acid, for example will convert a solid fat to a liquid fat, as the long-chain loses its flexibility and ability to intertwine with neighboring molecules and the resulting intermolecular forces are reduced. The reverse is true as well. If we hydrogenate unsaturated fats, we will make them more saturated and their consistency will change from liquid (like vegetable oils) to solid oils, like animal fats . You may have noticed margarine contains partially hydrogenated vegetable oils. By hydrogenating just the right amount, the liquid vegetable oils (like olive oil) can be made to have a consistency very similar to butter.
(same as the cracking reaction last step)
A more controlled kind of elimination reaction requires reacting the molecules with a base. The resulting reaction mechanism looks very much like the nucleophilic substitution reactions we saw already. there are two main types of such reactions, categorized the same way as we categorized the Nucleophilic substitution reactions. They are E1 and E2 reactions, representing first-order and second order, respectively, elimination reactions whereby a base (aka poor nucleophile) will participate in the reaction to remove two atoms from a substrate organic molecule, leaving a double bond in its place.
The base we use, just like the nucleophile in the SN1 and SN2 reactions, needs to have a lone pair that is can use to form a new bond with a hydrogen on a carbon adjacent to the carbon that had the leaving group LG attached.
17.3.3.1 E1 Reaction Mechanism
The following first-order E1 mechanism shows the rate determining step is the slow elimination of the leaving group, followed by the rapid removal of the beta hydrogen by the base and the formation of the double bond.
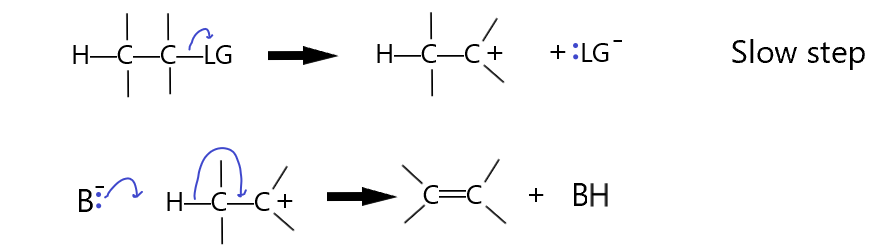
The E1 mechanism is more likely if the leaving group is what is called a good leaving group. Notice that the leaving group ends up with a lone pair. It is essentially also a base but it’s just a weaker base than the base we started with, B:–. The first step, the LG leaves, is the slow step. This step is simply the breaking of the C-LG bond to form the carbocation and the LG anion. Clearly, if LG is a better leaving group, a weaker base, the reaction will be favored. The second step is rapid by comparison as the carbocation produced in the first step is not very stable. The more stable this carbocation is, the faster this first step will happen because it will be more energetically favorable. The addition of electron donating groups to the carbon with the LG on it will make this reaction more likely, thus, an E1 reaction is more likely if the carbon with the LG on it is secondary or even tertiary. Additionally, the more heavily substituted carbons will also react more because the carbon changes from a tetrahedral (sp3) to a trigonal planar (sp2) configuration with the substituents having more room in the transition state. (less steric interaction internally). Substrate reactivity strength :
(CH3)3C—LG > (CH3)2CH—LG > CH3CH2—LG > CH3—LG.
Notice that the first step of an E1 reaction is the same as the first step of an SN1 reaction. So what is the difference? How do you know, if you are given a substrate with a good leaving group and a reactant which type the reaction will be? First of all, the leaving group must be a good leaving group, meaning it leaves of its own accord and does not need to be pushed off. But this is common to both SN1 and E1. If the other reactant is a good nucleophile, then it is a poor base, so it will react via SN1 but if the other reactant is a good base (weak electrophile) then the predominant mechanism will be E1, and a different product will ensue.
17.3.3.2 E2 Reaction Mechanism
If the LG is not a good leaving group it may need some help. This happens when the Base attacks the beta proton at the same time as the LG leaves. This single step mechanism involves two species in the slow step so it is a second order reaction, hence it is called an E2 reaction.
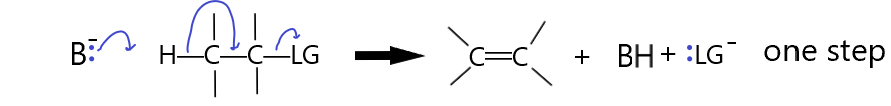
If the base is a better base than the LG is a leaving group, then the reaction may occur this way. Thus, very reactive bases (strong bases) will tend to force the reaction to occur via this E2 mechanism. Weaker bases will tend to result in the E1 mechanism above. The E1 mechanism resulted in an intermediate carbocation. If this carbocation is not stable, like in a primary carbon, then the E2 mechanism will be favored because there is no carbocation formation in the concerted mechanism. In the E1 mechanism, the tetrahedral carbons are converted to trigonal planar carbons. If the carbons are heavily substituted with R groups, the relaxing of the steric strain favours the reaction. Unlike SN2, where steric hindrance favors primary carbon attack, the base in an E2 reaction does not attack the α-carbon, it attacks the β-hydrogen and so a tertiary α-carbon will not cause steric hindrance. Thus, just like in an E1 reaction, the more substituted carbons are more likely to react than the less substituted ones. Substrate reactivity strength:
(CH3)3C—LG > (CH3)2CH—LG > CH3CH2—LG > CH3—LG.
17.4 Reactivity and influences
The reactivity of the components is affected by several factors, some of which compete with each other. We have mentioned several factors above. Let’s summarize here.
17.4.1 Electronegativity and inductive effects.
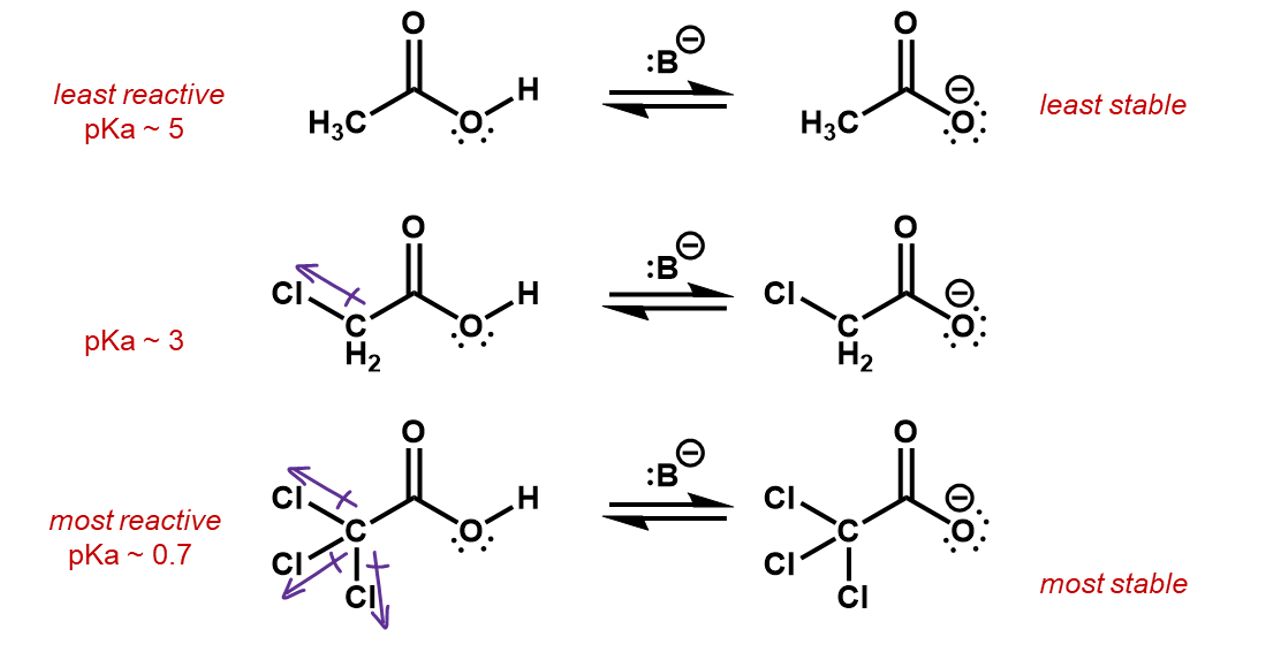
Electronegative elements tend to pull electron density towards them. This is called the inductive effect. It may seem counter-intuitive, but higher electronegative negative ions, actually result in lower nucleophilicity. This is because the higher electronegativity elements are more stable as an anion than less electronegative ions. Consider the following ions. Each has a lone pair to contribute to a nucleophilic reaction. The strongest nucleophile is the one with the lowest electronegative ion in it.
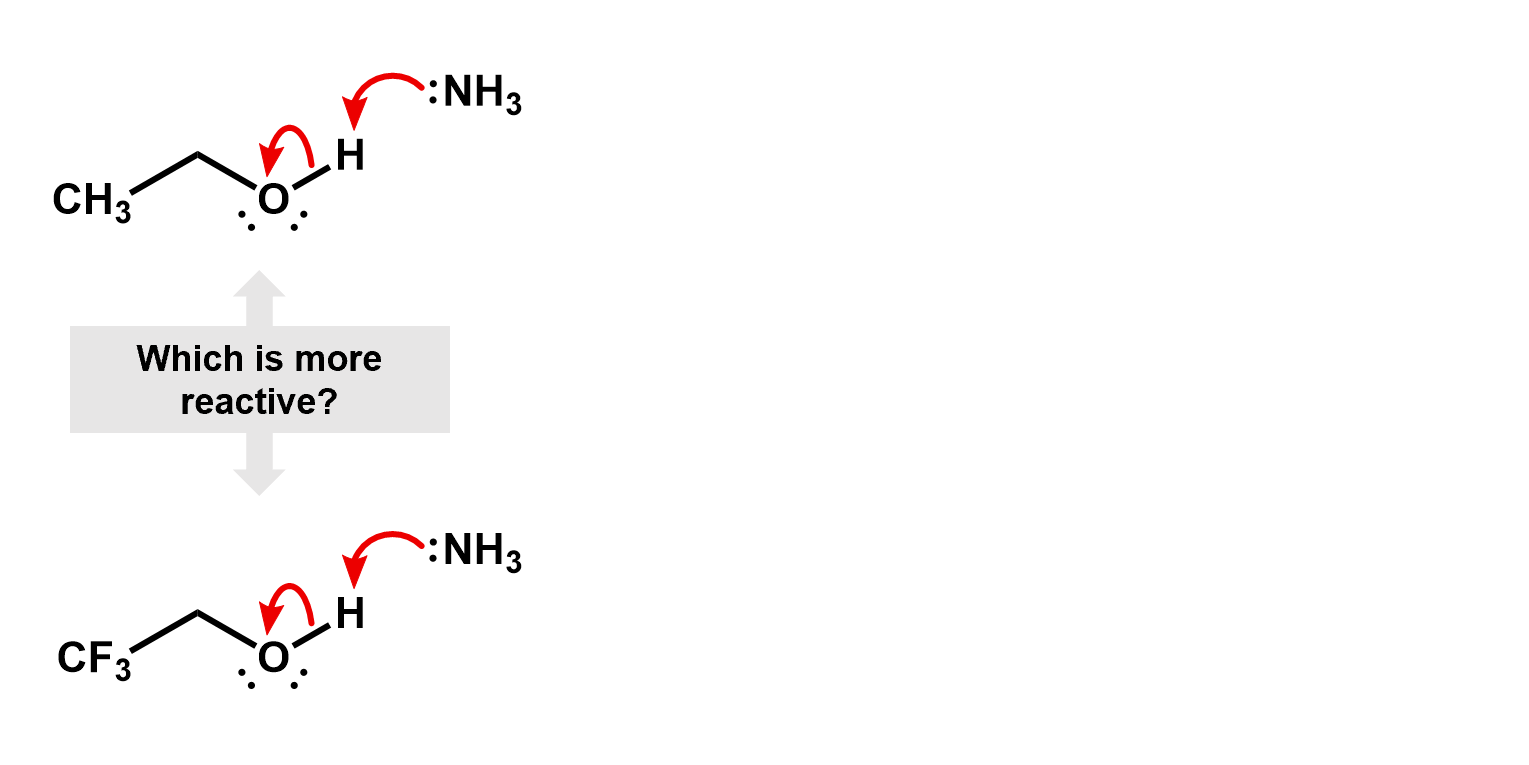
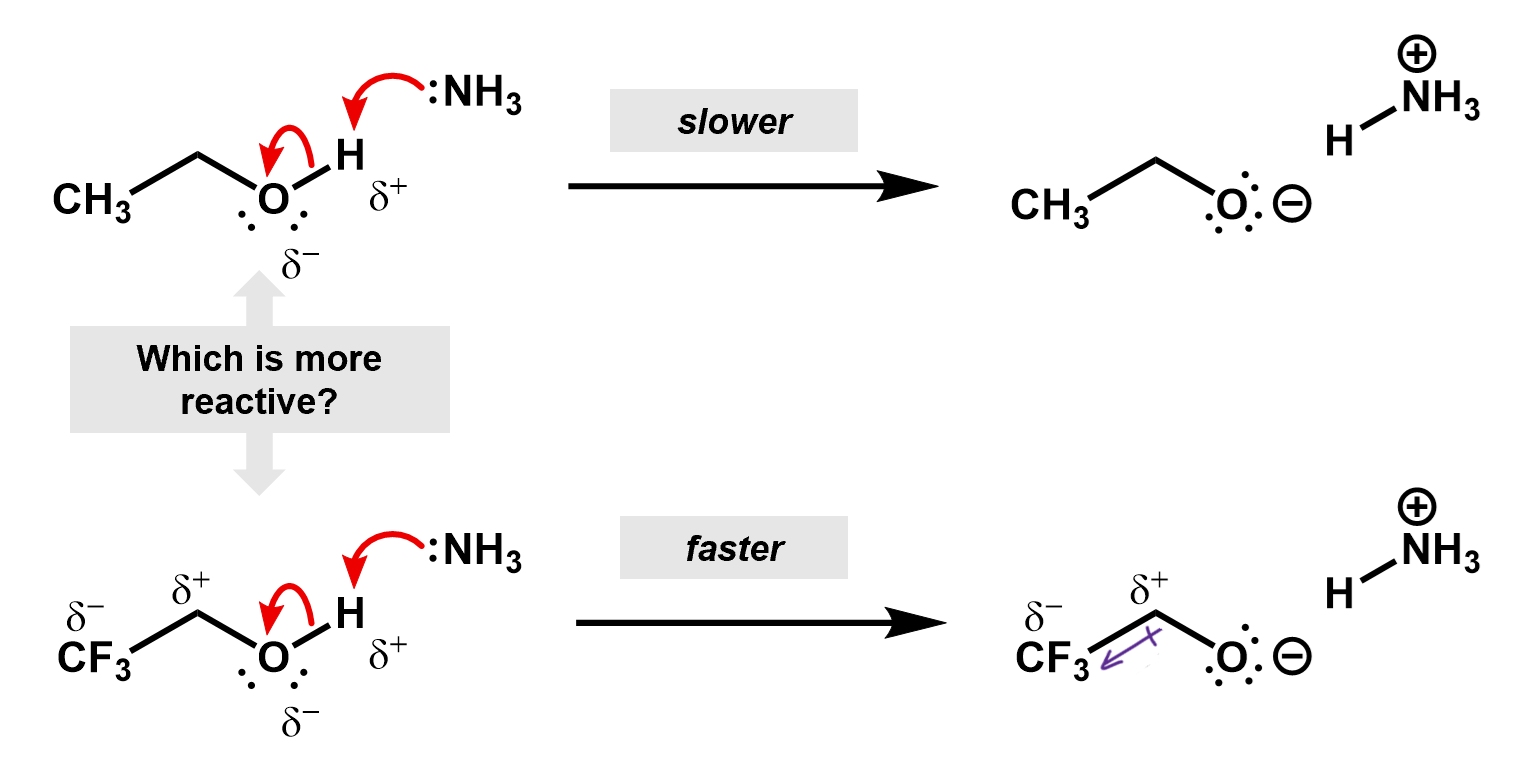
Thus, if we were to compare two reactions, one with fluorine and one without, we would be able to figure out which one is faster. Consider the deprotonation of a ethanol versus an ethanol with the beta carbon fully fluorinated.
The CF3 group is an electron withdrawing group, inductively pulling electron density off the rest of the carbon backbone, whereas, a CH3 group is actually an electron donor, pushing electron density onto the attached carbon. This is useful in the creation of a carbocation intermediate.
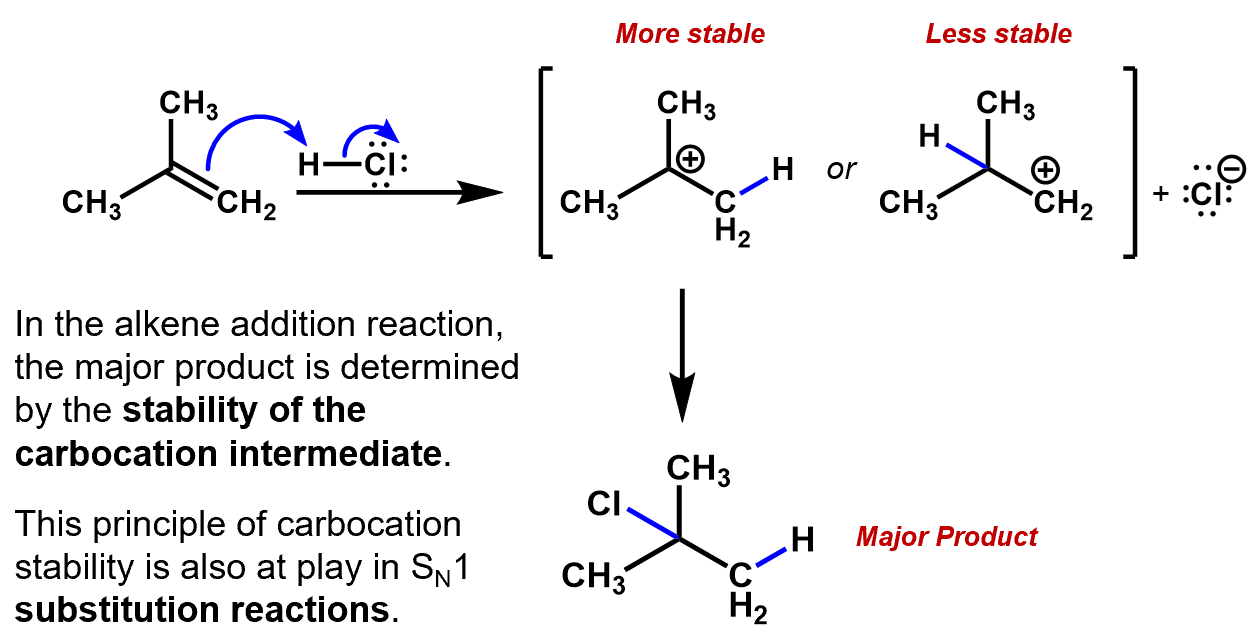
We have seen this in the discussion of the Markovnikov addition of HCl to 2-methylpropene.When a pair of π electrons (nucleophile) jumps to an electrophile (e.g. a proton), the newly formed bond to the proton could be attached to either carbon from the former double bond. The remaining carbon will end up with a positive charge. If one of those carbons has methyl groups on it, it will form the more stable carbocation, so it will become the more likely candidate for the final location of the Cl.
Which of the following acids is most reactive?
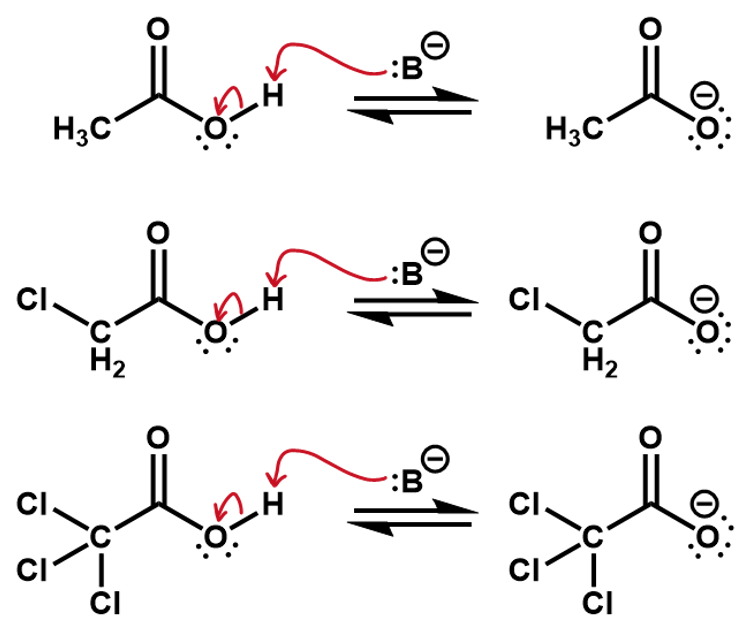
7.4.2 Atom Size and Polarizability
In a competition between two different nucleophiles, the more polarizable the nucleophile, the more reactive it is. Thus, For example, in an SN2 reaction of OH- and OS- on a bromomethane, the the latter will react faster because its more polarizable. The S atom, being larger has more room for its valence electrons and it more easily forms dipoles that can lead to the electron pair jumping onto the substrate.
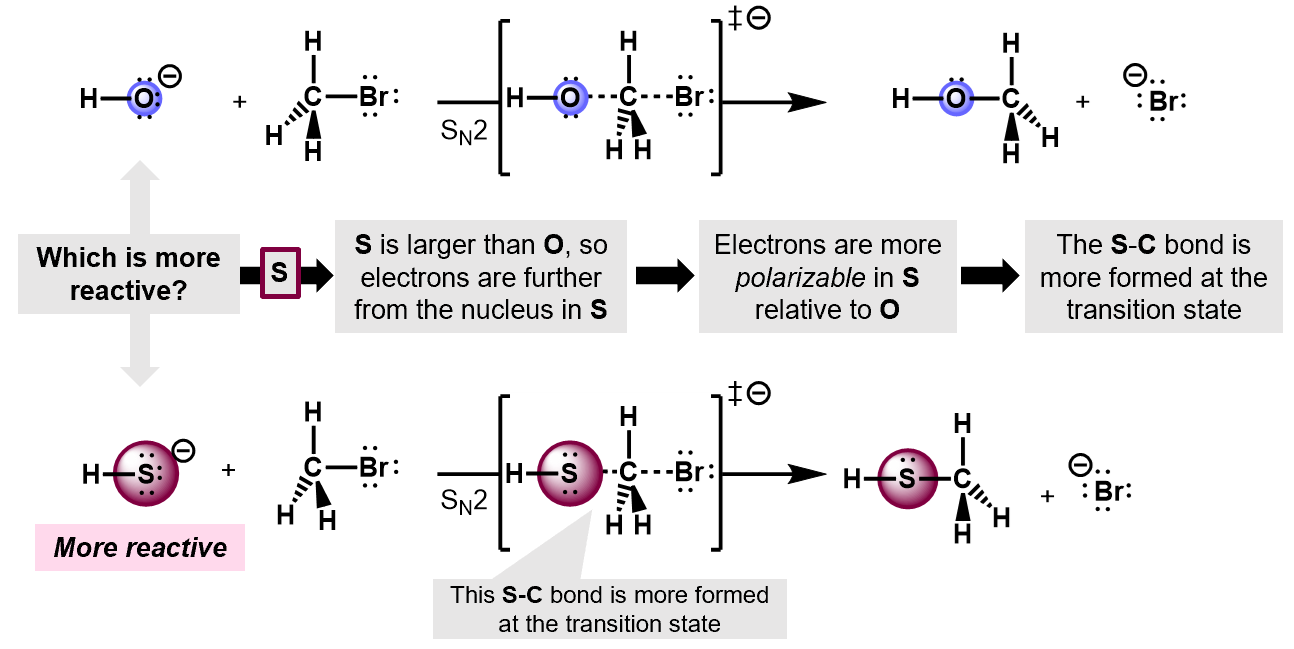
7.4.3 Delocalization effects.
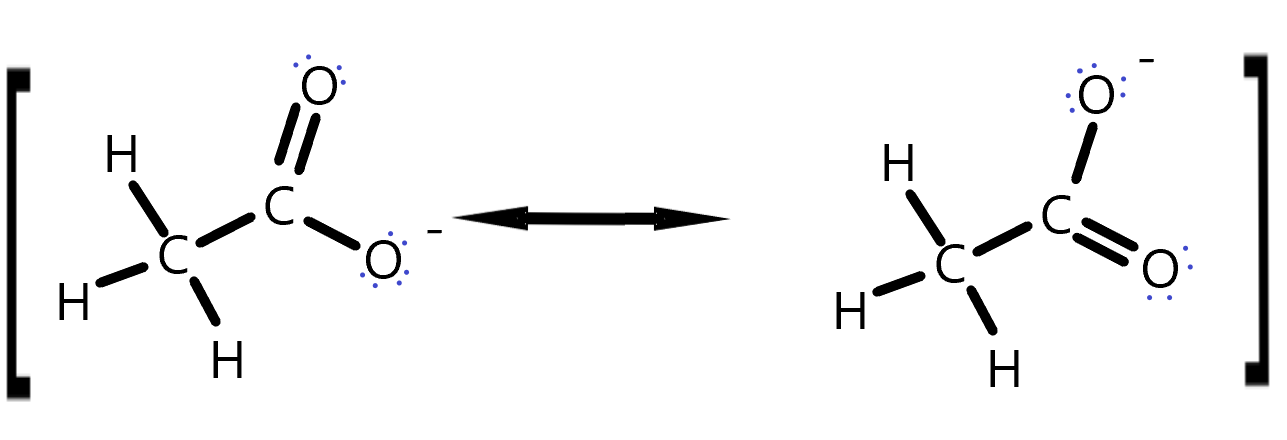
Charges that result from reactions can be stabilized by delocalization. We have seen how resonance structures can show that charges on atoms on average are not as high as one particular Lewis Dot structure might indicate. These resonance stabilizations can occur on any molecules that has a π system of electrons that can be moved. For example, the carboxylic acid group can stabilize the negative charge that is left behind when it is deprotonated. Thus, a proton on an alcohol -CH2OH, will be less acidic than a proton on a -COOH group. Try this for yourself.
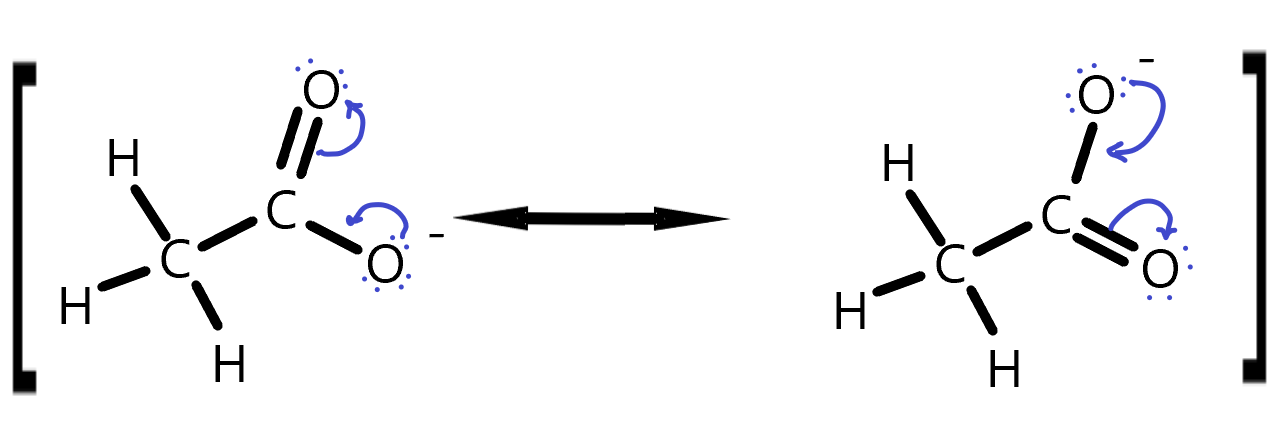
Draw curly arrows to represent the movement of electrons in this set of resonance structures. The square brackets here are meant to indicate this is really the same molecule but with different electron positions.
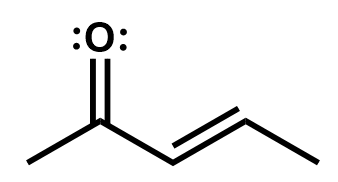
Delocalization via these resonance structures can be very useful to determine reactive sites on a larger system of π bonds, like you would find in an aromatic compound such as benzene.
Draw the resonance structures of this molecule to determine where the reactive sites (positive or negative) are.
7.4.4 Steric effect
Sometimes, the reactive site is hidden from other reactants by bulky substituents, either on the substrate or on the reactant.
R groups are much bulkier than simple hydrogen substituents on a carbon and make approach quite difficult for the nucleophile. The bulkier the nucleophile, the less the nucleophilicity.
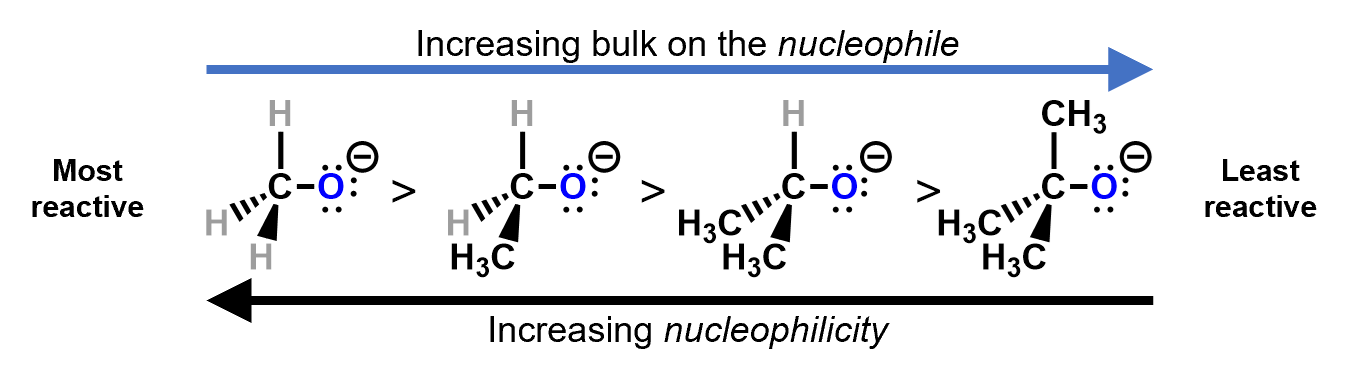
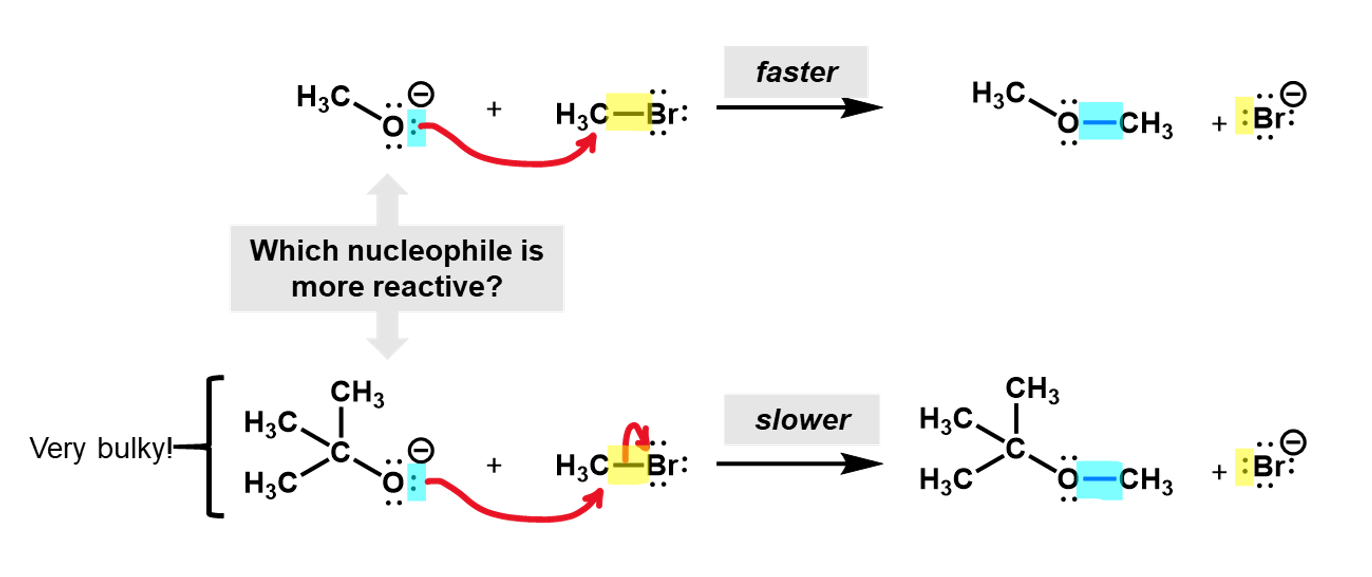
For example, consider a nucleophilic substitution reaction where different alkoxides react with bromomethane. Which of these two will be more reactive?
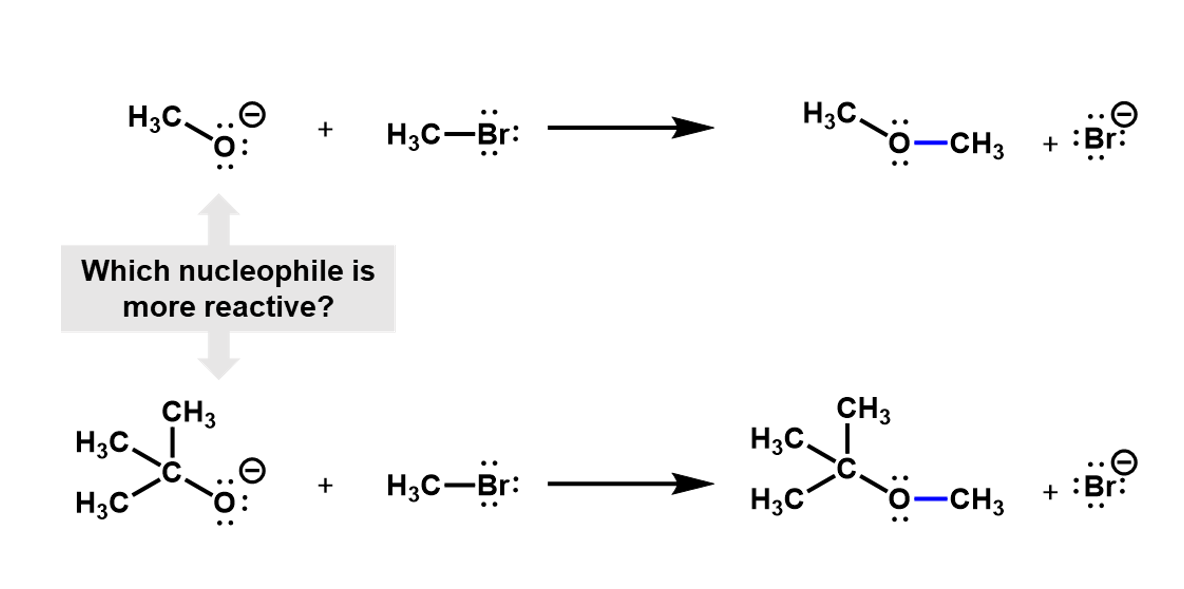
Looking at these two nucleophiles, we see that the tri-methyl substituted alcoxide is much more bulky and would hinder the approach of the bromomethyl, making it harder for the reaction to occur
Before we practice more organic chemistry, here is a guide to help you analyze a reaction and its mechanism:
- Housekeeping
- Redraw starting materials in a useful way (draw lone pairs, bonds, etc.)
- Number the (relevant) atoms
- Find reactivity
- Determine bonds formed and/or broken
- Identify the nucleophile and electrophile
- Draw the mechanism or product
- Use curved arrows
- Double check formal charges, atoms, total charge are conserved
End.
Practice:
Now try these practice exercises. Don’t look at the answers until you have given it a good effort.
Practice 2: Draw the mechanism using curly arrows.
What kind of reaction is this?
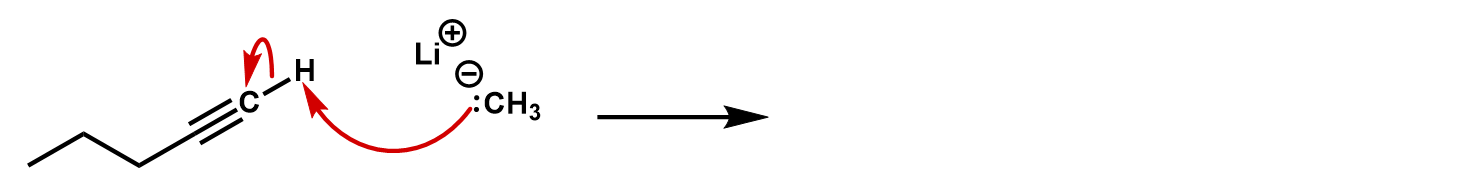
Practice 3: Identify the product of the following and identify the kind of reaction.
Practice 4: Finish the reaction. What kind of reaction is this?
Extra Organic Reactions Questions for practice.
Answer to reactivity question.
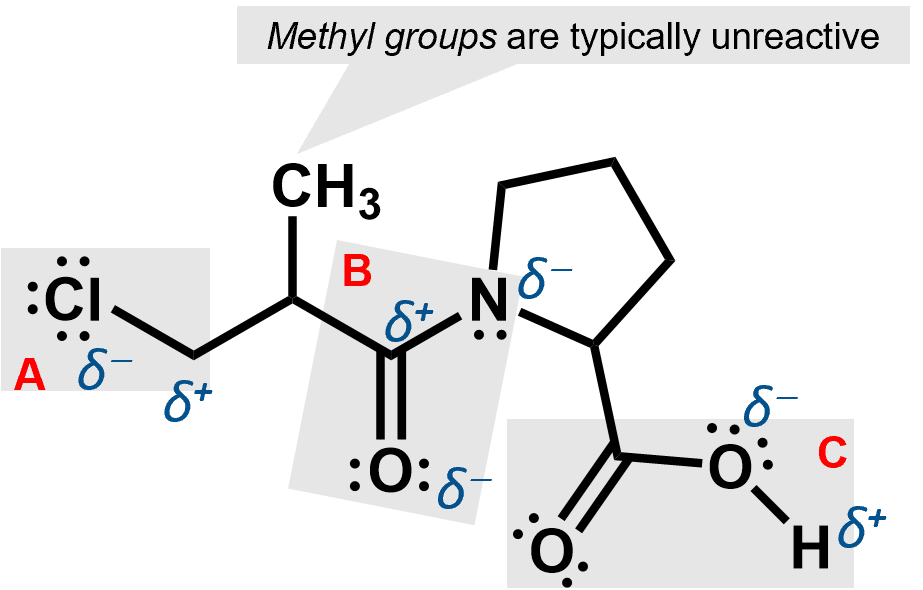 The three functional groups here are shaded. A is an alkyl chloride, B is an amide and C is a carboxylic acid group. Each of them can cause partial charges to appear because they involve atoms that are more electronegative than the carbons to which they are attached.
The three functional groups here are shaded. A is an alkyl chloride, B is an amide and C is a carboxylic acid group. Each of them can cause partial charges to appear because they involve atoms that are more electronegative than the carbons to which they are attached.
First, identify what happens. Are any bonds broken or formed?
Then draw in the information necessary to complete the reaction. This may mean you have to redraw the molecules to show things that were hidden like bonds and lone pairs.
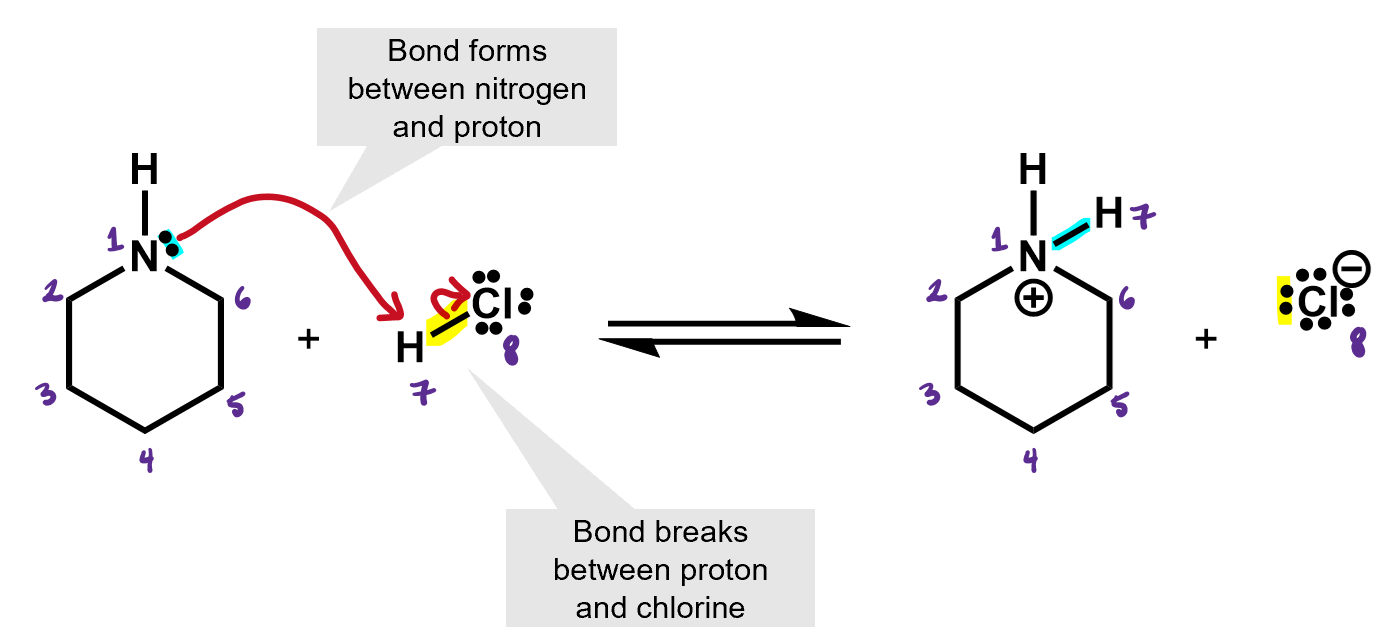
What kind of reaction is this? It’s an acid base reaction. A proton was exchanged.
Practice answer 3 what is the product formed here?
First identify what is happening. Where any bonds broken or formed?
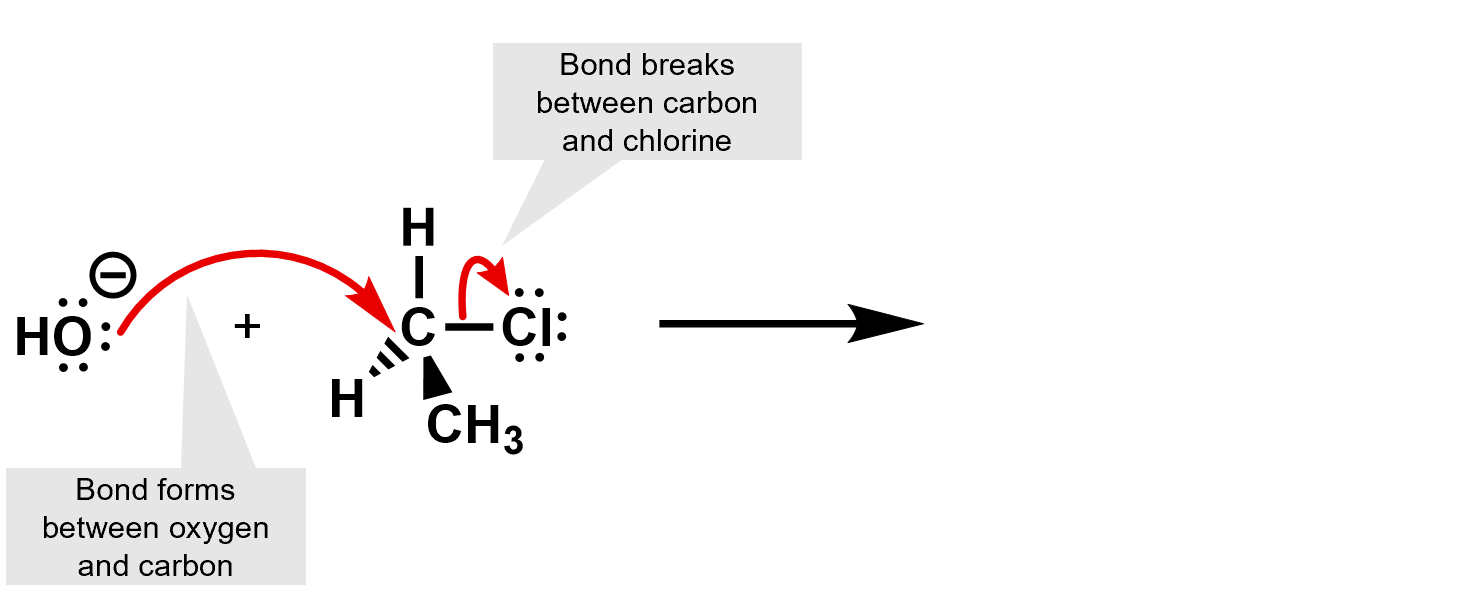
Then use the curly arrows to complete the reaction and determine the products.
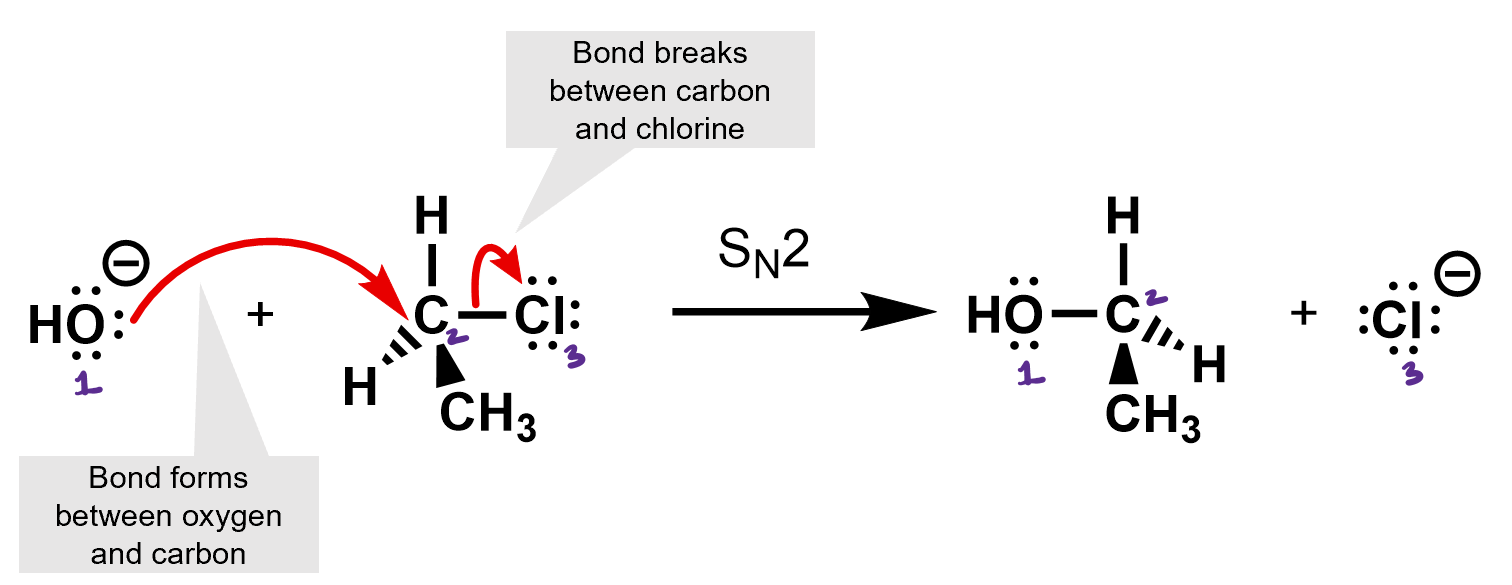
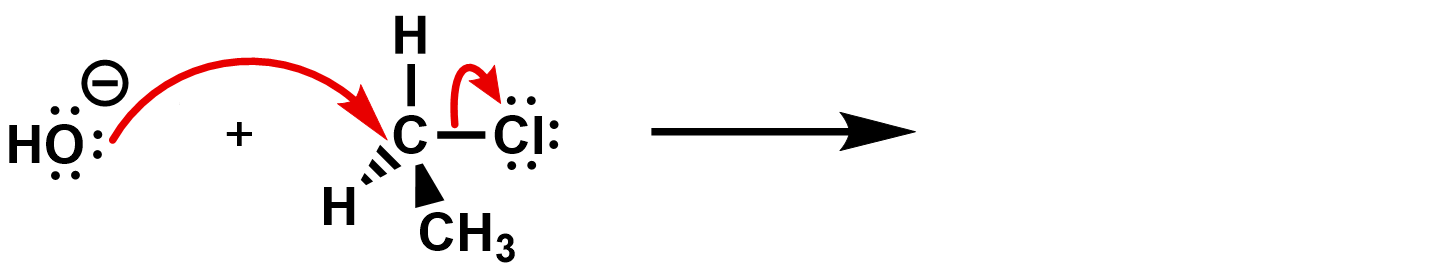
What kind of reaction is this? It’s an SN2 reaction. The hydroxide is the nucleophile, and the chloride is the leaving group.
First, identify what is happening.
Then complete the reaction.
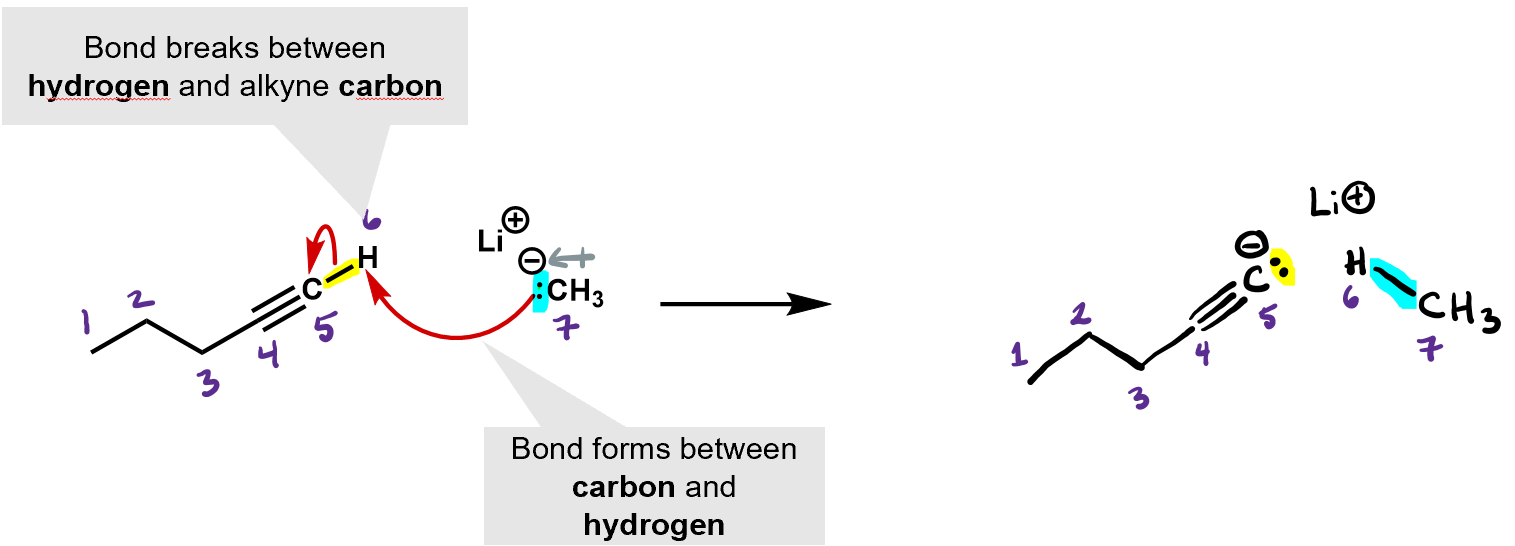
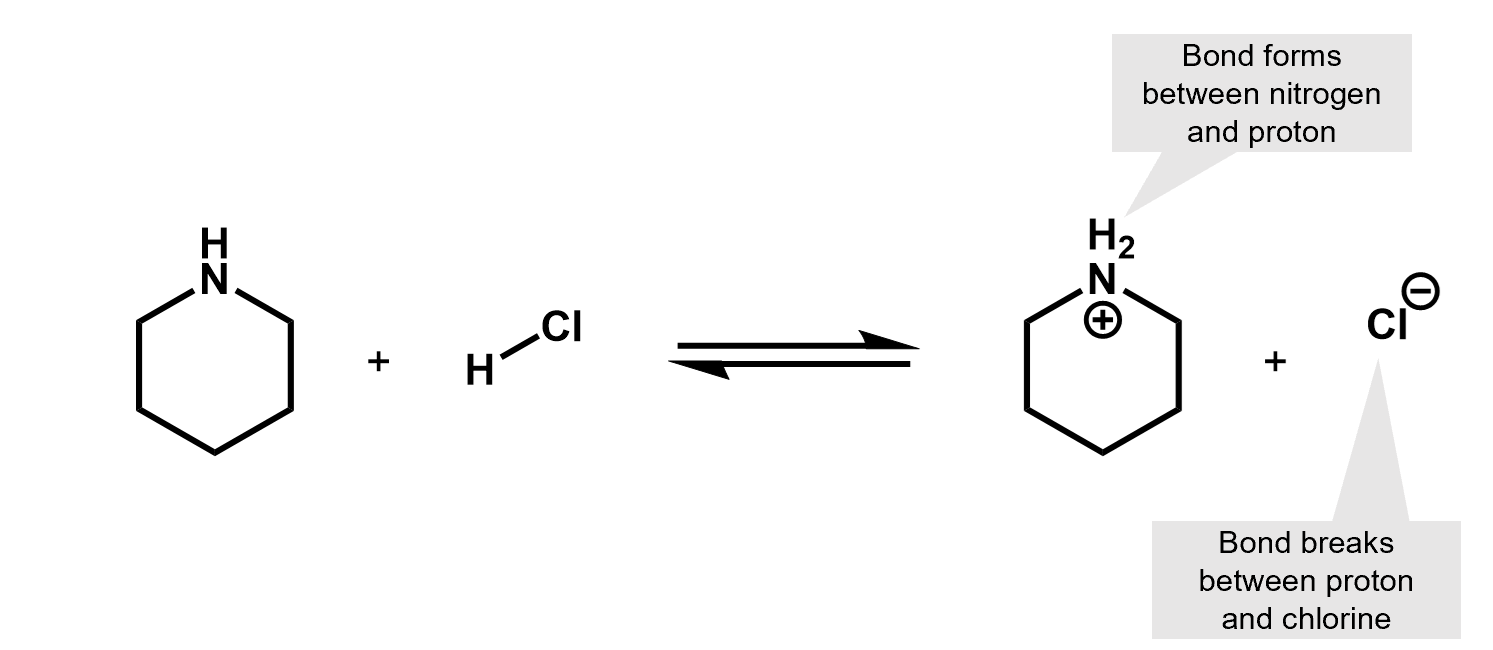
What kind of reaction is this? It’s an acid base reaction. A proton was exchanged.
First, look for places where the charges build up. Do these charges make the reaction more or less likely?

In this case, the fluorines pull electron density away from the oxygen making it more favorable for the O-H bonding pair to jump onto it. The inductive effect stabilizes the oxygen anion.
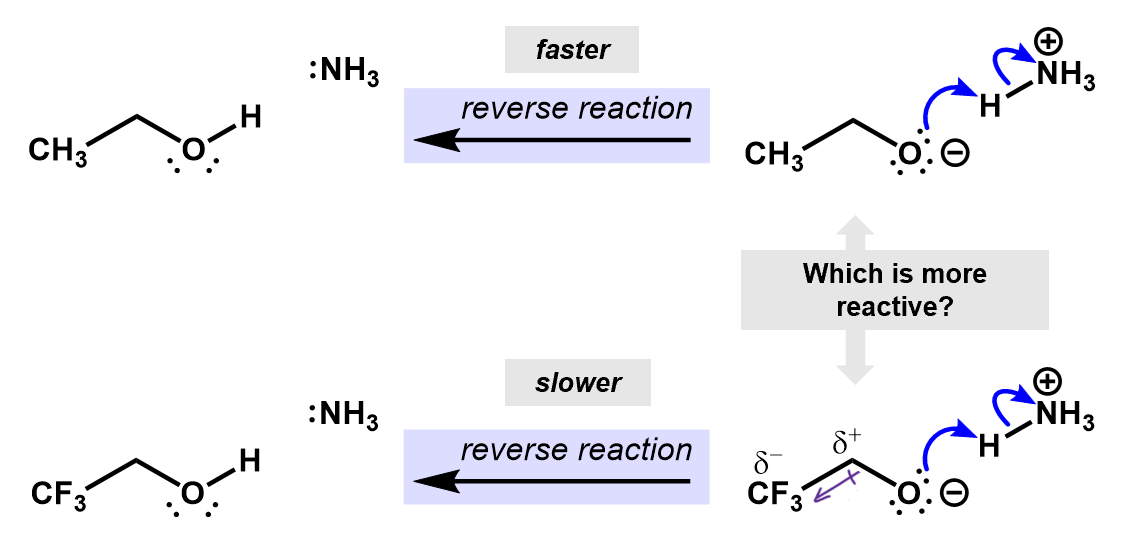
If we consider the reverse reactions to these, the inductive effects can also be used to predict the relative speed of reaction. This time, the fluorines are helping to slow down the reverse process. The induced charges from the CH3 group are stabilizing the oxygen anion, making it less reactive.
Ethanol Resonance answer. The lone pair electrons jump from the negatively charged oxygen to make a double bond and simultaneously, the other oxygen’s π-bonding electrons with the C jump onto the O, making it negative. This distributes the negative charge between the two oxygens on a time average.
Practice Inductive effect answer
The chloro substituents are electron withdrawing so in effect, the whole CCl3 group will withdraw electron density off the carboxylic group, stabilizing the negative charge more than the CH2Cl group and more than the CH3 group, which is in fact electron donating.
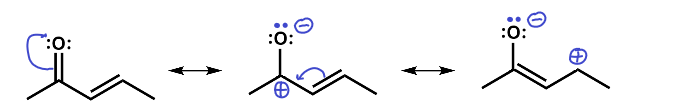
Here we see that there are three resonance structures. Note that not all are equal probability but all are possible. There are two carbons that end up with a partial positive charge as a result of this resonance and so they could act as electrophiles in a reaction. The oxygen with its partial negative charge and lone pairs could act as a nucleophile in a reaction.
You might have been tempted to move the π electron pair the other way. That would have produced a highly improbable entity with a negative carbon and a positively-charged electron-deficient oxygen bonded together. Oxygen is highly electronegative and this structure therefore is highly improbable.
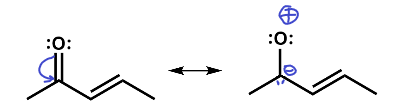
This structure is wrong.
- The α-carbon is the one to which the leaving group is attached originally and to which the nucleophile will be attached afterwards. It's the carbon closest to the reaction. A β-carbon would be a carbon attached to the alpha carbon. It's one carbon away from the reaction site. You will see this kind of terminology often used. ↵
