5 Energy, Enthalpy and Thermochemistry
Michael Mombourquette
5.1: Introduction
Thermodynamics is the study of heat (thermo) transfer (dynamics). Actually, we will be looking at how energy is transformed from one location to another, how it exchanges between heat and work, how to account for the gain or loss of energy and its relationship to chemical reaction energy changes. We will be focusing on energy measurements (calorimetry) and (mostly) the First-law of Thermodynamics and its applications.
To begin the study of the transformation of energy in chemical (or other) processes, we need to first develop a few terms that we will use. These definitions must be very tightly specified and understood in order to properly follow some of the more complex logic in later pages.
5.1.1: Definitions
- Thermodynamics
- The science of transformation of energy.
- Energy
- The capacity to do work or to transfer heat
- Work
- Work is one means of transferring energy from our system to the surroundings. Work is given the symbol
 in equations. Work has several aspects to it. There is mechanical work, electrical work, light-energy work and work of expansion or contraction of a gas (
in equations. Work has several aspects to it. There is mechanical work, electrical work, light-energy work and work of expansion or contraction of a gas ( work). In chemistry, we generally are only interested in work and will use the symbol to refer only to that type of work. All other types of work will be referred to using a prime on the symbol,
work). In chemistry, we generally are only interested in work and will use the symbol to refer only to that type of work. All other types of work will be referred to using a prime on the symbol,  .
.
One of the most common definitions for work is when a force  displaces an object by a distance
displaces an object by a distance  does work
does work  . (also known as
. (also known as  ). This is a mechanical work and serves as the basis for our definition of work.
). This is a mechanical work and serves as the basis for our definition of work.
Work can also be done by pushing back the atmosphere (piston in a cylinder). In this case, one pushes back against the atmospheric pressure  and moves the piston a distance . The force needed to push back the atmosphere is actually
and moves the piston a distance . The force needed to push back the atmosphere is actually  where
where  is the area of the piston. Hence, we can calculate the magnitude of the work done as
is the area of the piston. Hence, we can calculate the magnitude of the work done as
![\[|w| = P_{atm}\times A \times \Delta x = P_{atm} \times \Delta V\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-ddff8eb7e07ae6c4db68e06223314952_l3.png "Rendered by QuickLaTeX.com")
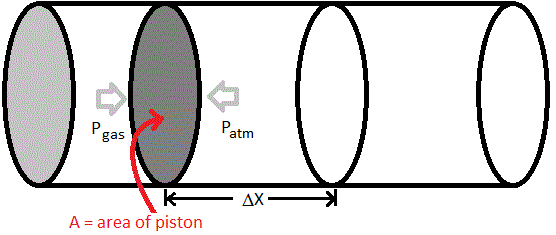
We call this atmospheric work. The only thing left to decide is the sign convention. (Described later) Since the system increased in volume and hence, did the work of pushing back the atmosphere (positive value for ΔV), it has used up energy so we can now write down a more complete definition for PV work as:
w = -Patm × ΔV
In chemical situations, we are normally interested only in the atmospheric work done when gases are evolved or used up in the reaction.
| both of these can be forms of work and of heat energy | KE = 1/2 mv2 PE = mgh lifting mass m by height h against acceleration due to gravity, g. Since mg is the ‘force” of gravity and  is really a distance, we can write. is really a distance, we can write.PE = f × d force times distance. |
Work is organized energy, lifting a book, etc.
Heat (defined later) is random (disorganized) energy that is released or absorbed, often as a result of some work being done, e.g., sound (energy) dissipates through air, when a book drops and hits the floor and the floor gets warmer (molecules vibrate more rapidly) spreading out from the spot where the hit occurred. Thus, the work done to raise the book (increasing its potential energy) is converted to kinetic energy upon release and then to thermal energy (energy is released as heat) as the book strikes the floor.
One can also transfer energy into or out of a sample via light-energy and electrical work
(We include this type of work in our catchall term w’ ).
- SI units of energy (work or heat):
- Let’s start with work: w has dimension of force times distance.
The SI units are N m = kg m2 s-2 = Joules (J). These are the same units used for heat transfer. - Heat
- Heat is the Energy transferred between the system and the surroundings as a result of a temperature differential. This is the only kind of energy transfer that does not involve doing work. Heat is given the symbol q in equations.
5.2: First Law of Thermodynamics
The First Law of Thermodynamics is often called the Law of conservation of energy:
- Energy can neither be created nor destroyed but only changed from one form to another.
OR
- The energy of the universe is constant.
OR
- The energy of a system which is isolated from its surroundings is constant.
We need a few more definitions so we can discus thermodynamics more easily.
- System
- The system is something we define ourselves and that definition then lets us write the equations we need and know what the symbols in the equations mean. The system is generally defined to be whatever we are interested in. That might be: the chemicals in a reaction, the reaction mixture, the chemicals and their container, etc. Essentially, we want to define the system in a way that makes the calculations as simple as possible.
- Surroundings
- After we have defined the system, the surroundings are defined by default. It is most simply defined to be the rest of the universe. Although, in practical experiments, we often refer to the surroundings as the rest of the room we’re in, or perhaps the rest of the chemical apparatus. For example, a large water bath surrounding a reaction chamber might be considered the surroundings if it is insulated from the rest of the universe.
- Closed System
- Any system that is sealed but not insulated, for example, an aluminum water bottle will not lose water but it will get warm as the heat from the day is absorbed into the bottle.
- Thus, Heat may transfer into or out of the system but no material can transfer.
- Isolated system
- An isolated system is both sealed against material transfer and insulated against heat transfer. For example, a thermos bottle is sealed so you don’t lose the contents and also insulated so the contents stay cold (or hot).
- Thus, no heat or matter can transfer between the system and its surroundings.
- Open system
- An open system can transfer both material and heat, for example, a paper coffee cup loses both water (as vapour) and heat to the surroundings.
- Insulated system
- An insulated system is designed so it does not lose heat to the surroundings or vice versa but it is not necessarily sealed against material transfer. For example, a Styrofoam coffee cup might lose material but will retain heat.
Note that the experimental reality may not quite match up with the definitions we use here. For example, an insulated system will still lose or gain heat because no amount of insulation is perfect. However, we can often consider the amount to be negligible as long as the rate of heat loss is very slow compared to the time scale of the experiment we have carried out. For example, it may take 30 minutes or so for the contents of a coffee cup to cool down to room temperature from the boiling point of water, so the cup is not a perfect insulator. However, we might still use a coffee cup as an insulated container for a thermodynamics experiment that we can complete in a few seconds. We will consider the coffee cup to be a perfectly insulated container in this experiment as long as the amount of heat lost over the short time of the experiment is quite small compared to the heat exchange involved in the experiment itself.
Energy transfer can be done in one of two ways:
- Work w can be done on the system by the surroundings (or vice versa). It can take the form of mechanical work or of electrical energy transfer.
- Heat q flows from the system to the surroundings (or vice versa)
There is a general sign convention that chemists use when describing q and w. When energy flows into the system as a result of heat or work, the sign is positive (the system gains energy). When energy flows out of a system, the sign is negative (the system loses energy. Both q and w refer, not to an amount of energy, but to an amount of energy transferred as a result of the process.
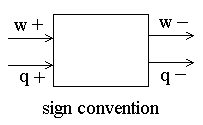 (note the sign convention)
(note the sign convention)
5.3: Enthalpy
For the initial part of this chapter, we will be looking at a specific type of case where the system undergoing the change is held at constant pressure. This, in fact, represents the way the majority of reactions are done in our chemical experience.
We define a term called enthalpy  is the energy transferred between a system and the surroundings under constant pressure. We cannot measure the absolute enthalpy of a system but we can measure the change in enthalpy for a process. If the Initial and final states have enthalpies
is the energy transferred between a system and the surroundings under constant pressure. We cannot measure the absolute enthalpy of a system but we can measure the change in enthalpy for a process. If the Initial and final states have enthalpies  and
and  , respectively then the change in enthalpy for the process can be defined as
, respectively then the change in enthalpy for the process can be defined as
![\[\Delta H = H_f - H_i\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-c646332c6491223a50607e7b4356b4f4_l3.png "Rendered by QuickLaTeX.com")
This definition is only good if H is a state function, i.e., the change in enthalpy depends only on the initial and final states, not on the process itself.
If we simply measure the heat evolved (or absorbed) during a process, we will not have a measure of the change in enthalpy since, in general, q is not a state function. It depends on how the process occurs.
We need to restrict our process measurements to specific conditions in order to be able to measure enthalpy directly. In this case, if we maintain constant pressure and then simply measure the heat transferred as a result of the process qp we will have a measure of the change in enthalpy. The subscript p refers to the fact that this heat was measured with the system held at constant pressure conditions.
Thus, we have simply,
ΔH = qp
the heat measured at constant pressure.
For chemical reactions we can write
ΔH = ∑pHp – ∑rHr
where subscripts p and r refer to products and reactants, respectively. If ΔH is negative, then there is a net flow of heat from chemical system to surroundings (heat is released during the reaction). This kind of process is called Exothermic (EXO sounds like exit).
If ΔH is positive, then net flow of heat from surroundings to chemical system (heat absorbed). Endothermic (ENDO sounds like enter)
| CH4(g) + 2O2(g) |
ΔH = -890 kJ/mol (EXOTHERMIC) |
| H2(g) + I2(g) |
ΔH = 52.2 kJ/mol (ENDOTHERMIC) |
Note that any time you see units of “/mol” on a thermodynamic parameter like ΔH, it means “per mole of reaction as written”. For example, in the methane combustion reaction above, for every mole of reaction, there is one mole of methane used up and two moles of oxygen used up. If you rewrote the reaction to have only one mole of oxygen and a half mole of methane
1/2CH4(g) + O2(g) → 1/2CO2(g) + H2O(l),
then the ΔH value would be half as much because the equation written that way has half as much chemicals in it.
5.4: Heat Capacity
One of the easiest ways to measure a heat transfer is to measure a change in temperature and then calculate from that, the heat transferred. We use the concept of heat capacity to do this. Heat capacity of a substance describes the amount of heat that can be absorbed by the substance for a given unit rise in temperature (unit = 1 K). It can be expressed in general as:
![\[C = \frac{q}{\Delta T}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-39b0aae5b3f8bd230764f64a754583dc_l3.png "Rendered by QuickLaTeX.com")
The Heat capacity can be expressed as a function of the amount of material. The heat capacity per mole is called the molar heat capacity (units J K-1mol-1). The heat capacity per gram is called the specific heat capacity (or just specific heat) (units J K-1g-1). It can simply be the capacity of the unit object. For example, a calorimeter (made up of or containing several materials, container, insulation, water, etc.) will have a heat capacity unique to it.
It is not completely correct to talk of heat capacity in terms of q since q is not a state function. We are better off using the state functions ΔH rather than q.
We assume that no electrical or mechanical work w’ is being done.
At constant pressure we measure heat as ΔH = qp and hence the heat capacity we need to use is Cp , where we define:
![\[C_p = \frac{\Delta H}{\Delta T}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-cd2406933e29eaf19cbc135e9befc996_l3.png "Rendered by QuickLaTeX.com")
Thus, we can write (at constant P)
ΔH = CpΔT
The units of Cp are J/K or J/℃ (remember that ΔT is the same whether it’s measured in ℃ or in K). Sometimes, we tabulate heat capacities in per mole or per gram values. If this is the case, we need to multiply the molar heat capacity by the number of moles or the gram heat capacity (specific heat) by the number of grams to get the total heat capacity. In these cases, we can think of modifying our equation for enthalpy to be
ΔH = nCpΔT
(for molar heat capacity values[1])
OR
ΔH = mCpΔT (for specific heat values[2])
Obviously, these equations hold Cp to be independent of temperature. In reality, while that’s a reasonable estimate for our purposes, it is not strictly correct. The heat capacity of any substance changes slightly with changes in temperature.
5.5: Thermochemistry (Calorimetry)
Now, Let’s quickly review a few definitions we will need to use in this section.
- Calorimetry
- Study of heat absorbed or evolved in chemical reactions
- Calorimeter
- Device used in Calorimetry to measure heat processes (normally thermally insulated from the surroundings)
- Heat Capacity
- Amount of heat to raise an object or a given amount of substance by 1℃ (1K).
- Molar Heat Capacity
- Amount of heat to raise one mole of a substance by 1℃ (1K).
(water has molar heat capacity of 75.4 J K-1 mol-1 . - Specific Heat
- Amount of heat required to raise one gram by 1℃ (1K).
(water has specific heat of 1 cal K-1 g-1 or 4.18 J K-1 g-1
Let’s try an example: A sample of 50. mL of a 0.20 M solution of HCl was mixed with 50. mL of 0.20 M NaOH in a coffee cup calorimeter. The initial temperature of both solutions was 22.2℃. After mixing, the temperature rose to 23.5℃. What is the enthalpy change for the neutralization reaction which occurred?
To start, we need to define the system. Since this case involves a reaction in a liquid solution where the chemicals are intimately involved with the solvent (solvation in water), we cannot really separate the water from the chemicals. We will use the chemicals and water as the system. The insulated cup is the boundary between the system and the surroundings and we will assume that the cup itself absorbs no heat and that no heat passes through the cup to the surroundings.
We have thus, a two step process to deal with.
- The reaction occurs, warming the system. Since no heat was evolved to the surroundings (coffee cup insulation prevented it) the value of q and of ΔH are zero since w is negligibly small (no volume change in a liquid). For that matter, ΔU is zero too (See Internal Energy at the end of this section).
- The system must be returned to the starting temperature by extracting heat from it or adding heat to it. Thus, the ΔT we calculate is of the step, after the reaction is over, where the system is returned to its original state.
H3O+(aq) + OH–(aq) 2 H2O (l)
Total volume = 100. mL, dilute aqueous so we assume density = 1.00 g/mL
mass of solution: msol = 100. mL × 1.00 g/mL = 100. g
For the cooling process:
ΔT = Tf – Ti = 22.2℃ – 23.5℃ = -1.3℃ (= -1.3 K)
The temperature was lowered from its high value back down to its starting point. Thus, we expect a negative temperature change. Another way of viewing this: We removed heat from the system to return it to its starting point. Hence, q and ΔH are negative (the reaction is exothermic).
This temperature change is happening to the system.
qp = CpΔT = mCm ΔT (Cm is specific heat, i.e., heat capacity per gram)
NOTE: to figure out which equation you need simply look at the units of the quantities you have and figure out how to cancel out the undesired units by the correct combination of C with the values you have. In this case, we have
qp = 4.18 J/K·g × 100. g × -1.3 K = -540. J
# mol H3O+ = # mol HCl = 0.50 L × 0.20 mol/L = 0.010 mol.
ΔH’ = qp = -540 J
Note that the ΔH shown here is for the whole reaction amount. It is not a molar amount; hence, the prime on the ΔH’. I use the prime here to distinguish the global quantity and no prime to show a molar quantity.
![\[\Delta H = \dfrac{1}{\frac{-540 \mathrm{J}}{0.010\mathrm{ mol}}} = -54. \mathrm{kJ/mol\;H}_3\mathrm{O}^+\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-d8b6a45a05bbd2c69fa96e7658715316_l3.png "Rendered by QuickLaTeX.com")
We’ll use the stoichiometry from the balanced chemical equation to convert from moles of H3O+ to moles of reaction. According to the equation
H3O+(aq) + OH–(aq) ![]() 2 H2O(l),
2 H2O(l),
there is one hydronium ion for every one reaction. So, we convert to moles of equation as follows:
![\[ \Delta H = \[\begin{array}{|c|c|} 540\;\mathrm{ J} & 1\; \mathrm{H}_3\mathrm{O}^+ \\ \hline \mathrm{0.010 mol H}_3\mathrm{O}^+& \mathrm{1 equation}\end{array} = -54.\mathrm{ kJ/mol equation}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-79aa2c31ca660d25baa9f1cc426cd73f_l3.png "Rendered by QuickLaTeX.com")
The value reported for the enthalpy change must always be accompanied by the balanced chemical reaction we used in the determination.
If we had balanced the chemical equation differently, say, to show one mole of water, it might look like:
1/2 H3O+(aq) + 1/2 OH–(aq) H2O(l)
In this balanced equation, there is one half a hydronium ion for every equation thus, our conversion would be
![\[ \Delta H = \[\begin{array}{|c|c|} 540\;\mathrm{J} & 1/2\; \mathrm{H}_3\mathrm{O}^+ \\ \hline \mathrm{0.010 mol H}_3\mathrm{O}^+& \mathrm{1 equation}\end{array} = -27.\mathrm{ kJ/mol equation}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-083dc5b5cc65dea663c4ff3b3e3ac044_l3.png "Rendered by QuickLaTeX.com")
Our test calorimeter is not that accurate (significant heat loss to the surroundings). The true ΔH for the first reaction is -56.02 kJ/mol and is valid for the heat of neutralization of any strong acid with a strong base in water.
Here is an example where we are able to isolate the chemicals separately as the system and directly measure the heat removed from the system.
0.510 g of ethanol is burned in a flame calorimeter containing 1200 g of water. The water is initially at 22.46℃ and is warmed up to 25.52℃ as a result of the reaction. What is the ΔH for one mole of ethanol?
In this case, we can more easily separate the gas reaction mixture from the water in the calorimeter which absorbs (all) the heat from the reaction. Therefore, we can use the chemicals themselves as the system. In this case, the insulation in the calorimeter serves to isolate a small portion of the surroundings (the water) so we can measure its temperature change directly, while ignoring the rest of the surroundings.
C2H5OH(l) + 3 O2(g) 2CO2(g) + 3 H2O(l)
![\[\Delta T_{water} = 25.52\Celsius - 22.46\Celsius = 3.06\Celsius = 3.06 K\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-f515f39ebfff59971d35b4937dad9cbd_l3.png "Rendered by QuickLaTeX.com")
We have a specified mass of water so we will start with that and look up and use a heat capacity which has grams (specific heat) to cancel out g and leave the desired units of energy.
![\[q_\mathrm{surroundings}=\underset{\mathrm{net\;heat\;capacity}}{\underbrace{\frac{1200.\;\mathrm{g}\;\mathrm{H}_2\mathrm{O}}\;{\textstyle\left|{\displaystyle\frac{4.18\;\mathrm{J}}{\mathrm{k}\;\mathrm{g}\;\mathrm{H}_2\mathrm{O}}}\right|}}}\;\;\underset{\mathrm{Temp\;change}}{\underbrace{3.06\;\mathrm{K}}}=15.3\;\mathrm{kJ}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-1e6c9ea729dc85ed04d0e5f060eee79c_l3.png "Rendered by QuickLaTeX.com")
This is the total heat absorbed by the water (surroundings) and is the negative of the heat evolved by the system. Hence,
![\[q_{p,system} = -q_{surroundings} = -15.3 kJ\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-227b70f3d926928b9176c920b6db72ce_l3.png "Rendered by QuickLaTeX.com")
Note that the q of the surroundings does not need to have a specification of constant P or constant V. It just doesn’t matter. Whatever energy is absorbed by the surroundings must have been given off by the system (First Law). Now, the system must be specified to be constant pressure if you need to equate the heat exchanged,  , to enthalpy change,
, to enthalpy change,  , of the system.
, of the system.
![]()
The other thing we need to do to calculate the enthalpy is to determine the heat per mole since enthalpies for a chemical reaction are always reported in units of kJ/mol of reaction.
![]()
Finally, we need to translate the enthalpy change into units of kJ/mol of equation as written, rather than kJ/mol of one particular chemical.
![\[\Delta H = \begin{array}{|c|c|} -1380\mathrm{ kJ} & \mathrm{1 ethanol} \\ \hline \mathrm{1 mol ethanol} & \mathrm{1 equation} \end{array} = -1380 \mathrm{ kJ/mol}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-fc19ec045b03e1481c16f65b41829651_l3.png "Rendered by QuickLaTeX.com")
It is important to note that this reaction is written for one mole of a reactant burned completely in oxygen. This type of reaction is called a combustion reaction (test)and the ΔH is called a heat of combustion. We could simply tabulate ΔcombH (ethanol) and know that the reaction is as written above.
Generally, we cannot use or report a enthalpy change for a chemical reaction unless we have also specified the chemical reaction. Had we written the reaction with different coefficients (still balanced) then the enthalpy change would have been different. Consider the reaction above written as follows.
1/3 C2H5OH(l) + O2(g) 2/3 CO2(g) + H2O(l)
This way, we have specified the reaction per mole of H2O produced. The value for ΔH for this reaction is 1/3 that for the reaction written for one mole of ethanol.
Thus, the enthalpy change for this reaction can be calculated, starting again from the enthalpy change for one mole of ethanol
![\[\Delta H = \begin{array}{|c|c|} -1380\mathrm{ kJ} & \mathrm{1/3 ethanol} \\ \hline \mathrm{1 mol ethanol} & \mathrm{1 equation} \end{array} = -460 \mathrm{ kJ/mol}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-c7094289e0b30eddaeef99c64f9cf2de_l3.png "Rendered by QuickLaTeX.com")
This is the enthalpy change for the reaction written above with only one mole of water produced.
5.6: Standard Enthalpy Changes
Reaction energies depend on the conditions under which they were measured. In order to be able to record energies and tabulate them in a way that others can make sense of them, we define a standard state. This standard state is then used to define the state of both the reactants (before reaction) and products (after reaction). Recall that since ΔH and ΔU do not depend on the path, only the initial and final state. Hence this is a useful thing to do.
Thermodynamic Standard state should not be confused with the standard conditions (STP) used for Ideal Gas calculations and tabulations. They aren’t the same.
Standard State:
Pressure = 100 kPa (1 bar)*
Concentration = 1 mol/L, (solutions)
[Temperature = for tabulation purposes only, normally but not exclusively 25℃ ]
What are elements in their standard states? How do we decide which ones to use?
- Take hydrogen:
- Most hydrogen exists at standard conditions as H2(g) [On earth, minute amounts may exist as H(g). There is far more hydrogen in space, where the atomic form predominates]. We choose the most common form on earth at normal (standard) conditions, H2(g).
- Now try Carbon:
- Obviously, carbon is solid at Std Conditions but do we choose graphite, diamond, Bucky balls (Buckminster Fullerenes)? Graphite is the defined standard, it is more a common and more stable allotrope than diamond or Bucky balls.
So how do we specify standard state in general? Normally, we just have to be careful to write the correct phase of the element or compound for the appropriate temperature. The following is a list of elements and compounds and the proper standard state at two different temperatures
| Substance | T=25℃ (298.15K) | T=100℃ | T=120℃ |
| hydrogen | H2(g) | H2(g) | H2(g) |
| oxygen | O2(g) | O2(g) | O2(g) |
| carbon | C(s,graphite) | C(s,graphite) | C(s,graphite) |
| water* | H2O( ) ) |
H2O() or H2O(g) |
H2O(g) |
| methane | CH4(g) | CH4(g) | CH4(g) |
| pentane (bp=36℃) | C5H15() |
C5H15(g) | C5H15(g) |
| methanol dissolved in water** | CH3OH(aq) | CH3OH(aq) | CH3OH(g) |
* We see that since water exists in two different standard, stable forms at 100℃ and P=1 bar, that there are two different possible states we can indicate, both of which are ‘standard’ states at 100℃. The problem here would be that the difference between them would be the difference in energy (enthalpy) of vaporization. Be careful of the states specified.
** The final entry in the table shows methanol, a molecule that can dissolve in water. At or below 100℃, it is in standard state if the concentration is 1 mol/L. Any other concentration means not standard state. Note that above 100℃, water doesn’t exist in liquid form at standard conditions so methanol would only exist in gas form at p=1 bar alongside the water, which would also have to be at 1 bar if it were to be claimed to be standard state.
We’ll see later that a simpler definition for standard state is “activity = 1″
Temperature is not part of the definition. It is merely the experimental temperature for which most of the results are tabulated. We indicate Thermodynamic quantities measured at standard conditions by using the superscript zero as in ΔH° (pronounced delta H not)
eg. CH4(g) + 2 O2(g) → CO2(g) + 2 H2O(l) ΔrH° = -890.4 kJ/mol
NOTE: When we see the superscript ‘not’ on the symbol ΔrH° it means that all the reactants and products must be in their defined standard state at a defined pressure of 1 bar (and/or concentration of 1 mol/L for solutes).
Defined Reaction Types
Recall that the value we write down for the enthalpy of a reaction depends on how we have written the balanced chemical equation for that reaction. Hence, we either need to include the complete balanced chemical reaction with our values of ΔrH° or we need to define a way of writing the equations so we don’t have to actually specify them. The latter is the way we do it in many cases. Two common defined reaction types and their associated enthalpy changes are given here.
- Standard Formation reactions are written by default such that exactly one mole of the compound of interest is formed from its constituent elements in their thermodynamic standard states. It is important to remember what the standard state of an element is when we try to write a particular standard formation reaction. For example, water if formed from hydrogen and oxygen. So the standard formation reaction for water is:
H2(g) + 1/2 O2(g) → H2O(l) ΔfH° = -286. kJ/mol.
See how the standard form of hydrogen and oxygen is diatomic gas while that of water is molecular liquid. This is because the normal temperature of T=25℃ (and standard P=1 bar) is used unless otherwise specified. For example, had we been seeking the enthalpy change for the same reaction at a temperature T=120℃ (and P=1bar), we would still have calculated a standard enthalpy of formation but the standard states would be different.
H2(g) + 1/2 O2(g) → H2O(g) ΔfH° = -327. kJ/mol
Note that value for the standard enthalpy change at this temperature is not the same as at room temperature because we had to incorporate the enthalpy of vaporization of ~41 kJ/mol for water. This is still a ‘standard enthalpy change’ because all the chemicals are at standard state for T = 120℃ and P = standard pressure of 1 bar.
Note also that we assumed the heat capacity of the products and reactants don’t contribute significantly to the overall enthalpy change for this reaction under the temperature change from 25℃ to 120℃. This is not necessarily good assumption as we’ll see later (See Temperature Change and Enthalpy).
- Standard Combustion Reactions involve the complete oxidation of one mole of the compound of interest to its fully oxidized products. The word standard means everything is in its standard state for the temperature specified.
C2H6(g) + 7/2 O2(g) 2 CO2(g) + 3 H2O(l) ΔcombH° = -1560.4 kJ/mol
Since both of these types of reactions are now well defined, we can re-create the chemical equation exactly knowing only the compound of interest and the type of reaction. Hence, the values for the standard enthalpy changes for these defined types of reactions can be tabulated with need to only specify the chemical of interest. Thus, the following two specifications are complete because the reaction type and the chemical for each is specified so we can duplicate the appropriate balanced chemical equation that goes with each enthalpy change value.
ΔfH°(H2O)= –286. kJ/mol}
ΔcombH°(C2H6) = –1560.4 kJ/mol}
(since T was not specified for these two values, you must assume T=25℃)
5.7 Hess’ Law
Since enthalpy change, ΔH, is a state function, we can go to products via several routes and the enthalpy change will still be the same.
Another way to say this is: the enthalpy change in a reaction does not depend on the reaction pathway. This allows us to define any path we choose to get from reactants to products and, as long as we keep track of the enthalpy changes for each step, we will be able to calculate the overall enthalpy change for the process.
Let’s look at the example of ethane burning in excess oxygen to produce carbon dioxide and water. We have two possibilities: we make a series of reactions (1,2,3) that gets us to the final products and keep track of the enthalpy changes for each step; we simply burn the ethane in a calorimeter and measure the enthalpy change directly (labelled T). These two possibilities are diagrammed below where the vertical axis represents enthalpy.
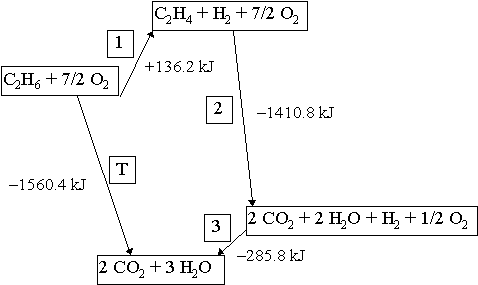
We can consider the reaction to occur in one complete process [T] or in three distinct steps [1], [2] and [3]. In either case, the total enthalpy change should be the same.
| [1] Ethane is heated to drive off H2. C2H6 |
ΔrH°1 = +136.2 kJ |
| [2] Ethene is burned in excess O2. C2H4 + 3 O2 → 2 CO2 + 2 H2O |
ΔrH°2 = -1410.8 kJ |
| [3] H2 is burned in excess O2. H2 + 1/2 O2 → H2O ——————————————– |
ΔrH°3 = -285.8 kJ |
| [T] overall reaction is sum [1]+[2]+[3] C2H6 + 3.5 O2 → 2 CO2 + 3 H2O |
ΔrH°T = -1560.4 kJ |
The example here may not seem to have been practical at first glance. After all, why set up three separate calorimetry experiments when we could have just done the overall experiment in one step? Well, it serves as a good demonstration of the principle. Additionally, we can use this idea to calculate enthalpies changes for reactions where direct measurement is impractical. Also, we can use this method to calculate the expected enthalpy change of a reaction using tabulated values without the need to do any experiments.
Example: Use the following standard enthalpies of combustion to calculate the standard enthalpy change for the formation of methane.
The standard formation reaction for methane is:
C(s) + 2 H2(g) → CH4(g) ΔrH° = ?
This will be our target reaction. We need to use the Standard Combustion Enthalpies listed here.
1)ΔcombH°(C) = -393.5 kJ
2)ΔcombH°(H2) = -285.8 kJ
3)ΔcombH°(CH4) = -890.4 kJ
Knowing the standard format for the combustion reaction (1 mol of reactant burns to completion in oxygen to produce fully oxidized products; CO2 and/or H2O in this case) we can easily write down the three reactions.
| 1) C(s) + O2(g) → CO2(g) | ΔrHº = -393.5 kJ/mol |
| 2) H2(g) + 1/2 O2(g) → H2O(g) | ΔrHº = -285.8 kJ/mol |
| 3) CH4(g) + 2 O2(g) → CO2(g) + 2H2O(l) | ΔrHº = -890.4 kJ/mol |
| Now lets add up the three reactions to give the target reaction | |
| 1) C(s) + O2(g) → CO2(g) | ΔrHº = -393.5 kJ/mol |
| 2) 2 ×[ H2(g) + 1/2 O2(g) H2O(g) | ΔrHº = -285.8 kJ/mol] |
| 3) CO2(g) + 2H2O(l) → CH4(g) + 2 O2(g) ———————————————— |
ΔrHº = +890.4 kJ/mol |
| C(s) + 2 H2(g) → CH4(g) | ΔrHº = -74.7 kJ/mol |
To get to the desired final reaction, we needed to use equation 2 twice (needed to use up 2 hydrogens). Thus, we also have added the enthalpy for that reaction twice (note the square brackets extends around the equation and the enthalpy value. We also reversed the direction of equation 3 (multiplied all coefficients by -1, and hence multiplied the enthalpy change by -1) Whatever we do to the equation, we must also do the ΔHº value.
5.8: Standard Enthalpies of Formation
The Standard Enthalpy of formation is defined as the enthalpy change of a standard formation reaction. We saw the definition of the reaction type earlier. Now let’s explore how we can use these enthalpy change values.
Here are a few reactions that can be defined as standard formation reactions and whose enthalpy changes are useable as standard enthalpies of formation.
| C(s,graphite) + O2(g) → CO2(g) | ΔfHº (CO2) = –393.5 kJ/mol |
| H2(g) + 1/2 O2 → H2O(l) | ΔfHº (H2O) = –285.8 kJ/mol |
| C(graphite) + 2H2(g) → CH4(g) | ΔfHº (CH4) = –74.7 kJ/mol |
Note that the phase of standard state carbon is solid graphite, not just solid. That’s because there are more than one solid phase of carbon, for example, diamond is solid carbon. In the cases where there are more than one allotrope of an element, the most stable one is defined to be the standard state.
Question: What will be the standard enthalpy of formation of an element in its standard state [like O2(g)]?
Answer: Zero. Consider the reaction that is being described in the question. It would look like this: O2(g) → O2(g). Clearly there is no reaction as the product and the reactant are the same so no enthalpy change has actually occurred. We can generalize this discussion to say: the standard enthalpy of formation of any element in its standard state is zero.
Now let’s use these with Hess’ law to determine the reaction enthalpy for the following reaction.
CH4(g) + 2 O2(g) → CO2(g) + 2H2O(l) ΔrHº1 (combustion of methane)
According to Hess’ law, we could choose to break down the reactants to their constituent elements in their standard state and then reconstruct them into the products.
| CH4(g) + 2 O2(g) → C(s) + 2H2(g) + 2O2(g) | ΔrHº2 |
| C(s) + 2H2(g) + 2O2(g) → CO2(g) + 2H2O(l) | ΔrHº3 |
From Hess’ law, we have, ΔrHº1 = ΔrHº2 + ΔrHº3
Let’s explore ΔrHº2 + ΔrHº3 a bit further.
In step 2, we broke the reactants (methane and oxygen) into their constituent elements at standard state. This is the reverse of the formation of these reactants so the total enthalpy change is simply the negative of the sum of the standard enthalpy of formation for the reactants (multiplied, of course by the appropriate coefficients to give us the correct stoichiometry of the desired reaction). Thus,
ΔrHº2 = – [sum of Std enthalpies of formation of reactants].
ΔrHº2 = ΣΔrHº (react)
In step 3, we took elements in their standard state and formed the products. Thus, the total enthalpy change is simply the sum of the standard enthalpy of formation for the products (multiplied, of course by the appropriate coefficients to give us the correct stoichiometry of the desired reaction). Thus,
ΔrHº3 = – [sum of Std enthalpies of formation of products].
ΔrHº3 = ∑ ΔrHº (prod)
So now, we can write:
For the combustion of methane we’re dealing with here, we can write
ΔrHº1 = [ΔfHº(CO2) + 2 ΔfHº(H2O)] – [ΔfHº(CH4) + 2 ΔfHº(O2)]
We can look up the values for Heat of formation in any standard reference book.
ΔrHº1 = [(-393.5 kJ) + 2 (-285.8 kJ)] – [(-74.7 kJ) + 2 (0)]
ΔrHº1 = -890.4 kJ
In general, we can write:
ΔrHº = ∑p ΔrHº(prod) – ∑rΔrHº(react)
Note that the coefficients n used here are unitless because they represent mole ratios, not numbers of moles. This allows the final enthalpy change values to have units of kJ/mol, as they must have. Note that if one or more of the reactants or products are not in their standard state, we simply can remove the superscript ‘not’ from the final enthalpy change symbol and the same equation will still work.
This procedure is merely a special application of Hess’ Law whereby we are adding the enthalpies of formation in a way that gives us the overall enthalpy change.
_____________________________________________________
Let’s try a few examples to get some practice with these calculations:
What is the standard enthalpy change for
4 NH3(g) + 5 O2(g) → 4NO(g) + 6 H2O(l)?
ΔrHº = ∑ ΔfHº(prod) – ∑ ΔfHº(reactants)
ΔrHº = [4 ΔfHº(NO) + 6 ΔfHº(H2O)] – [4 ΔfHº(NH3) + 5 ΔfHº(O2)]
ΔrHº = [4 (90.25 kJ/mol) + 6 (-285.83 kJ/mol)] – [4 (-46.11 kJ/mol) + 5 (0)]
ΔrHº = -1169.54 kJ/mol
Now here’s another one.
B5H9(g) reacts exothermically with O2 to form B2O3(s) and water. What is the standard enthalpy change for the reaction of 1 mol of B5H9(g)?
| balance this for 1 mol B5H9 |
B5H9(g) + O2(g) |
B2O3(s) + H2O(l) |
| 1 B5H9(g) + 6 O2(g) |
5/2 B2O3(s) + 9/2 H2O(l) |
ΔrHº = ΣΔfHº(prod) – ΣΔfHº(reactants)
ΔrHº = [5/2ΔfHº(B2O3) + 9/2 ΔfHº(H2O)] – [ΔfHº(B5H9) + 6 ΔfHº (O2)]
ΔrHº = [5/2 (-1272.77) + 9/2 (-285.83)] – [(73.2) + 6 (0)]
ΔrHº = -4541.4 kJ/mol
And another: 1 mol of benzene burns in air at standard state conditions and gives off 3267 kJ of heat. What is the enthalpy of formation of C6H6?
C6H6(‘) + 7.5O2(g) → 6 CO2(g) + 3 H2O(l)
| ΔcHº | = -3267 kJ/mol |
| ΔcHº | = ΣΔfHº(prod) – ΣΔfHº(reactants) |
| -3267 kJ/mol | = [6 ΔfHº (CO2,g) + 3 ΔfHº (H2O,l)] – [ΔfHº (C6H6,l) + 7.5 ΔfHº(O2,g)] |
| Substitute in values from data table. | |
| -3267 kJ/mol | = [6 (-393.5) + 3 (-285.83)] – [ΔfHº(C6H6,l) + 7.5 (0)] |
Solve for the only unknown:
ΔfHº (C6H6,l) = 49 kJ/mol
_______________________________________
Table: Standard heat of combustion and boiling point of some selected hydrocarbon fuels
![\[\begin{array}{llllc} \hline\\ \textrm{Substance}&\textrm{State}&\textrm{Heat of Combustion}&&\Textrm{Boiling Point}\\ &&kJ/mol&kJ/g&^{\circ}C\\ \hline \textrm{C carbon}&s&-393.5&-32.7& _\\ \mathrm{H_2\;hydrogen}&g&-285.8&-141.8&-253\\ \end{array}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-022999750a257088881ce1b1c1ceb2f8_l3.png "Rendered by QuickLaTeX.com")
5.9: Temperature Change and Enthalpy
The standard enthalpy data we find tabulated in thermodynamic tables is all for data at 25℃. This data may not be exactly the information we need. What if a reaction doesn’t actually happen at standard 25℃? We have two options: 1, we can assume that there is no significant difference in enthalpy change or, 2, we can calculate the effect.
In cases where there is only small temperature changes the former option may not be too bad. However, in some cases, especially where the temperature of reaction is far from 25℃ we should take the latter option.
Consider a reaction between chemicals A and B to produce C and D at some temperature T.
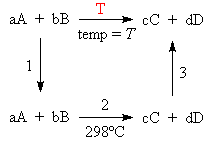
We could calculate the ΔH for the process 2 using standard thermodynamic data. Steps 1 and 2 involve temperature changes and we can calculate the ΔH for these steps if we know the heat capacities of the compounds involved. According to Hess’ law, the overall enthalpy change for the reaction at temperature T is the sum of the steps 1, 2 and 3.
| Step 1: | This is simply a temperature change and we can calculate the enthalpy change using the heat capacities of A and B. ΔH°1 = [a Cp° (A) + b Cp°(B)]ΔT1 (298 – T) |
||||||||
| Step 2: | ΔH2 = ΔHº298 (calculate from whatever means possible, for example you could use ΔfHº values.) |
||||||||
| Step 3: | Like step 1, this is simply a temperature change. Calculate the enthalpy change using the heat capacities of C and D. Note that the process is the reverse direction of step 1 and the sign of the temperature change is opposite.
ΔH3 = [c Cp°(C) + d Cp°(D)]ΔT3 (298 – T) NOTE: ΔT1= -ΔT3 |
||||||||
| Step T: |
|
Where have defined a new parameter ΔCp°.
![]()
The change in heat capacity (capacity of products – capacity of reactants) is actually slightly dependent on temperature and hence this formulation is not valid if the temperature T is very different from standard temp. (298 K). In most cases, the temperature effect is not extremely large but that is not general.
Like the general concepts involved in Hess’ Law, we simply add up the heats of the processes involved in order to determine the overall heat. This is merely another special application of Hess’ Law.
5.10: Internal Energy
Internal energy of the system is represented by the symbol U. It is a more general description of the energy of the system than enthalpy. We can never measure the absolute energy of a system but we can measure changes in the internal energy of the system using the state function
ΔH2U = Ufinal – Uinitial
Since we can never actually measure either Uinitial nor Ufinal we measure, instead, the energy transferred into or out of the system in the form of heat and work to obtain.
ΔU = q + w ( = heat transferred + work done)
This is, in fact, the mathematical expression of the First Law of Thermodynamics and is completely general, not relying on any of the restrictions we placed on enthalpy measurements. We can measure work and heat under any conditions to get the change in internal energy because it turns out that while neither q nor w are state functions, their sum is.
Example:
If 15 kJ of work is done by the surroundings on the system and the system loses 10 kJ of energy as heat to the surroundings then
| ΔU | = -10 kJ + 15 kJ |
| = +5 kJ |
Chemical reactions often involve changes in Volume (exp. gases)
| C3H8(g) + 5 O2(g) | → 3 CO2(g) + 4 H2O(g) | |
| 6 mol gas | 7 mol gas | → increase in volume at const. P |
If this reaction is to happen, it needs to push back the rest of universe (atmosphere) to make room for itself. Let’s put the reaction system into a cylindrical apparatus like that pictured at the beginning of this unit. As gas molecules are produced, the system expands to accommodate them and keep the pressure (inside and out) constantly the same. The amount of work done as the system expanded against constant external pressure can be calculated as:
|work| = force (a.k.a. pressure × area) × displacement (a.k.a. ΔX).
or |w| = P × A × ΔX or just P × ΔV
to fully define w, we need to recognize that if the system expands (does the work), the system lost energy so by definition, w is negative when volume expands (ΔV positive). So we can remove the magnitude formulation and put in a negative sign.
w = – PsurrΔV (gas expanding against an external pressure Psurr.)
ΔU = q + w = q – PsurrΔV
or qp = ΔU + Psurr ΔV
in this case, we can use the ideal gas law to get a more useful equation as long as the gas molecules all behave ideally.
qp = ΔU + ΔngRT.
which is also written as
ΔH = ΔU + ΔngRT.
We can use this formulation when the only volume increase comes from the fact that the number of moles of gas is different in products than in reactants. Note that the parameter ΔU is a value that is tied to the balanced chemical equation (just as we did for enthalpy changes). Thus, the value we used for ΔU and the value we used for Δng must be both for the same balanced chemical equation.
In a closed container (no volume change) there would be no work so
ΔV = 0 so: ΔU = qv.
subscript v refers to constant volume conditions.
Enthalpy is now better defined as
H = U + PV
Changes in enthalpy are
ΔH = ΔU + ΔPV since we normally work in a fixed pressure (atmosphere) situation, we can rewrite this as
ΔH =ΔU + PΔV.
To do these measurements under general conditions, we need to expand a few previous definitions. First, let’s look at heat capacity.
- At constant pressure, we measure heat as ΔH = qp, and hence the heat capacity we need to use is Cp, where we define
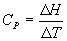
- At constant volume, the heat change we measure is ΔU = qv and the heat capacity Cv is defined as
-
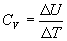
In many substances, the two heat capacities differ considerably. Consider one mole of ideal gas. We have
ΔH – ΔU = Δ(PV) = RΔT
![]()
Therefore: Cp – Cv = R for one mole of ideal gas.
Solids and liquids often have values of Cp and Cv that don’t differ much. On the other hand there is no consistent rule. Take for example water and benzene; for water,
Cp – Cv = R = 0.075R, while for benzene we have Cp – Cv = R = 5.1R.
Let’s do an example where we look at the PV work done.
When 2.00 mol of SO2(g) react completely with 1.00 mol O2(g) to form 2.00 mol of SO3(g) at 25℃ and constant pressure of 1.00 atm, 198 kJ of energy is released as heat. Calculate ΔU and ΔH for this reaction.
2 SO2(g) + 1 O2(g) → 2 SO3(g)
The heat released is |qp| since it was measured under constant pressure conditions. so we can quickly say
ΔH‘ = -198 kJ (negative sign since the heat is released, i.e., exothermic)
ΔH = -198 kJ/mol (trivial in this case since the molar quantities given exactly match the numbers in the equation)
ΔH’ = qp + w = qp – PΔV (total PV work)
or (since we have the molar values)
ΔU = ΔH – PΔV. (remember, this is the PV work per mole which just happens to equal the total PV work in this case)
Since we have constant P and T conditions, the only change in PΔV will be due to change in numbers of moles of gas molecules. We can use the stoichiometry from the equation as written to determine PΔV:
| ΔU =ΔH – ΔngRT | or | ΔH =ΔU + ΔngRT |
| Note: Either of these two equations above are valid for any situation where the T and P are constant but where the number of moles of gas molecules changes because of stoichiometry. Pick one and learn it. | ||
Δng = (#moles product gas – #moles reactant gas) per mole of equation
Δng = 2 – 3 = -1.
Note that the coefficients used here to calculate Δn are really unitless as they represent ratios of moles, not actual measures of moles.
| ΔU | = -198 kJ/mol + -(-1 × 8.31451 J K-1mol-1 × 298 K) |
| = -198 kJ/mol + 2.48 kJ/mol | |
| = -196 kJ/mol |
