13 Solubility
Michael Mombourquette
13.1: Introduction
We’ve seen many ionic compounds. Many are soluble in water and many are not. There is no simple set of rules which we can use to predict which ones will be soluble or not.
Some useful trends have been observed.
- multiple-charged ions are less soluble than single-charged ions.
- smaller ions are more soluble than larger ions.
For example,
- cations like NH4+ and all alkali metal ions are small, singly charged and are all soluble.
- anions like Cl–, Br–, I–, NO3– and ClO4– are all soluble except with Mg2+ and Ag+.
- Most hydroxides are insoluble (except with alkali metals as in NaOH or KOH).
- Salts of doubly charged ions CO32-, S2-, PO43- mostly are insoluble.
- Salts of singly charged versions of these are soluble (HCO3–, H2PO4– )
Reactions where soluble compounds react to form insoluble ones are called precipitation reactions. The reverse is a dissolution reaction where solid compounds dissolve upon addition into water.
For a table of solubilities click here
13.2: Solubility Product Constant
Since salts dissolve into electrically charged species (as do acids and bases) they fall in the general category called electrolytes. We saw in the case of acids and bases that some dissociate completely into ions and are called strong while others dissociate only partially and are called weak. This terminology applies to salts as well. A strong electrolyte is any electrolyte (acid, base, salt) that dissociates 100% into ions. A weak electrolyte sets up an equilibrium in water so that some exits as the original undissociated species and some exists as ions in solution or as undissolved solid.
In reactions involving ions which have the potential to form insoluble compounds, we must consider equilibrium conditions in order to decide whether or not a precipitate (ppt) will form.
for example, the reaction between the calcium cation and the sulphate anion is written as follows (always put the solid as the reactant and the ions as the product.)
CaSO4(s) ⇄ Ca2+(aq) + SO42-(aq)
Recall that the CaSO4 is a pure solid and therefore the activity is 1. Since this equilibrium constant is the product of the ion concentrations (solubility products) we use subscript sp for this new value.
Ksp = [Ca2+][SO42-]
Ksp is the solubility product constant and the right hand side of this equation is often called the ion product.
In general, for the reaction
AxBy(s) ⇄ X Am+(aq) + Y Bn-(aq)
we have
Ksp = [Am+]X[Bn-]Y @ equilibrium.
We can perform equilibrium type calculations as we did previously.
Example:
CaSO4 at 25℃ in water has a solubility of 4.9×10-3 M, i.e., enough CaSO4 dissolves in water to make a solution which has a nominal concentration of 4.9×10-3 M. Nominal meaning we pretend that the CaSO4 remains as a molecular unit in water when we quote this number. It doesn’t. What is its equilibrium constant Ksp?
| CaSO4(s) | ⇄ | Ca2+(aq) | + | SO42-(aq) | |
| I | solid, ignore | 0 | 0 | ||
| C | x dissolves | +x | +x | ||
| E | x | x |
x is the solubility, x = 4.9×10-3 M
Ksp = [Ca2+][SO42-] = x2 = (4.9×10-3)2 = 2.4×10-5.
In this next example, we will use a known Ksp value to determine the solubility of a salt.
The solubility product constant of PbCl2 is 1.7×10-5 at 25℃. What is the solubility of lead(II)chloride in water at 25℃?
| PbCl2(s) | ⇄ | Pb2+(aq) | + | 2 Cl–(aq) | |
| I | solid, ignore | 0 | 0 | ||
| C | x dissolves | +x | +2x | ||
| E | x | 2x |
| Ksp | = [Pb2+][Cl–]2 |
| = x(2x)2 = 4x3 = 1.7×10-5 | |
| x3 | = (1.7×10-5)/4 |
| x | = (4.3×10-6)1/3 |
| x | = 1.6×10-2 . Solubility = 1.6×10-2 M |
In this next example, we are going to use equilibrium ideas to determine if a precipitate will form.
Let’s mix equal volumes of 2.0×10-4 M AgNO3 with 2.0×10-4 M NaCl. Does a ppt form?
Since equal volumes of the two solutions are added together, the volume doubles and hence, each concentration is halved.
The only possible reaction between the four ions present here Ag+, Na+, Cl–, NO3–, is:
AgCl(s) ⇄ Ag+(aq) + Cl–(aq) (NaNO3 is very soluble.)
Q = [Ag+][Cl–] = (1.0×10-4)(1.0×10-4) = 1.0 × 10-8.
Ksp = 1.7×10-10. thus, Q > K therefore (rxn will proceed to left) ppt will form.
13.3: Common Ion Effect
The solubility of an ionic compound in a solution which already contains one of the ions in that compound is reduced. This is the common ion effect.
Consider the following.
PbCl2(s) ⇄ Pb2+(aq) + 2 Cl–(aq)
If we add some NaCl (or any other soluble chloride) we cause a stress on the equilibrium ([Cl–] increases). LP states that the equilibrium will shift to the left to try to use up the extra chloride, i.e., more ppt will form.
Example
What is the solubility of PbCl2 in 1.00 M HCl?
| PbCl2(s) | ⇄ | Pb2+(aq) | + | 2 Cl–(aq) | |
| I | solid, ignore | 0 | 1.00 | ||
| C | x dissolves | +x | +2x | ||
| E | x | 1.00+2x |
Ksp = [Pb2+][Cl–]2 = 1.7×10-5.
x (1.00 + 2x)2 = 1.7×10-5.
Now let’s use the small K approximation to simplify this and avoid using the quadratic equation to solve this. Since K is small, we assume x is small with respect to the initial amounts. Thus we assume that 1.00 + 2x can be approximated by just 1.00. Thus…
x (1.00 M)2 = 1.7×10-5.
x = 1.7×10-5 M.
If no HCl or any other chloride had been present we would have determined the solubility to be 1.6×10-2 M. The solubility is significantly reduced due to common ion effect.
The common ion effect is a very useful way to deliberately reduce the solubility of slightly soluble ions. Consider Radon Ra2+(aq). This radioactive ion can be separated out of a mixture by adding an anion which will precipitate out with it such as the sulfate ion.
RaSO4 (s) ⇄ Ra2+(aq) + SO42-(aq) Ksp = [Ra2+][SO42-] = 3.6 × 10-11.
solubility of RaSO4 in pure water is (3.6 × 10-11)1/2 or 6.0×10-6M.
If we add enough H2SO4 to make a 1.00 M solution. we find
[Ra2+] = Ksp/[SO42-] = (3.6 × 10-11)/ 1.00 = 3.6 × 10-11M.
13.4: Effect of pH on Solubility
Many weakly soluble ionic compounds have solubilities which depend on the pH of the solution. A direct example is hydroxides since the OH– ion is directly involved in the equilibrium constant. Other cases of pH dependence may not be quite so simple.
NOTE: pH is commonly defined as -log[H+]. This is not accurate. It should more accurately be defined as -log[aH+], i.e., activity of H+ should be used. Click here for more info.
Example
Zinc hydroxide Zn(OH)2 has Ksp=4.5×10-17
In pure water:
Solubility is computed as follows:
| Zn(OH)2(s) | ⇄ | Zn2+(aq) | + | 2 OH–(aq) | |
| I | solid, ignore | 0 | 0 | ||
| C | x dissolves | +x | +2x | ||
| E | x | 2x |
Ksp = 4.5×10-17 = x(2x)2.
x = (4.5×10-17 )/4]1/3 = 2.2×10-6 M.
the resulting pH is [OH–] = 2x = 4.4×10-6 M therefore
pH = 14 – pOH
= 14 – (-log( 4.4×10-6))
= 8.64
This pH is the equilibrium pH resulting from dissolving the zinc hydroxide in pure water. LP states if we stress the system (by changing the pH) then the equilibrium will shift to reduce the stress.
if pH < 8.64 (more acidic) then [OH–] decreases (rx shifts right to try to produce more). Solubility increases.
if pH > 8.64 (more basic) then [OH–] increases (rx shifts left to try to use more). Solubility decreases.
Now what will happen in this example is we start again but this time, we try to dissolve the zinc hydroxide into a solution buffered at pH = 6.0.
[OH–] = antilog[ -(14 – pH)]
= antilog[ -(14 – 6)] = 1.0×10-8.
Ksp = [Zn2+][OH–]2
[Zn2+] = Ksp/[OH–]2 = 4.5×10-17/(1.0×10-8)2 = 0.45 M. (= x).
Much larger solubility than in pure water.
Here’s a point! Tooth enamel is largely Ca5(PO4)3OH (hydroxyapatite). What do you think will happen to your teeth if organic acids (say from the partial digestion of foods rich in sugar by the saliva and biota in your mouth) are allowed to stay in your mouth. (brush your teeth within 15 minutes of eating even one mouthful). Similarly, drinking carbonated (carbonic acid) drinks are instantly dangerous to your teeth. The more of them you drink, the less tooth enamel you have and there is no natural process for building up your teeth once you degrade them.
| H2CO3/HCO3– | pKa=6.4 |
| HCO3–/CO32– | pKa=10.3 |
Any salt containing acidic or basic ions will have Solubilities that depend on pH. Take for example, CaCO3. If the pH is sufficiently low the HCO3–/CO32– reaction will completely use up the carbonate. In the distribution diagram for the carbonic acid system below, we see that at pH=8 there is (nearly) no carbonate (blue) left.
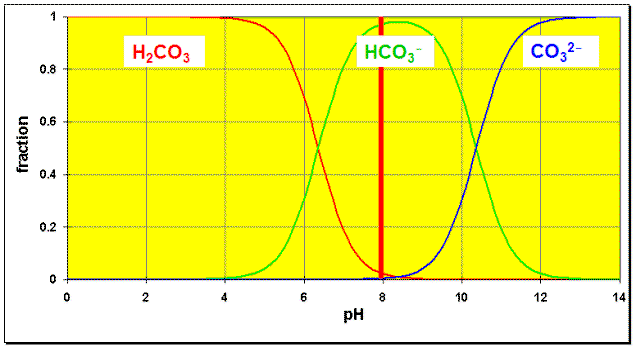
| (1) | CaCO3(s) | ⇄ | Ca2+(aq) + CO32-(aq) | Ksp(CaCO3)=3.36×10–9 |
| (2) | CO32-(aq) + H3O+(aq) | ⇄ | HCO3–(aq) + H2O(l) | 1/Ka2(H2CO3)=1/4.7×10–11 = 2.2×1010 |
| Sum | CaCO3(s) + H3O+(aq) | ⇄ | Ca2+(aq) + HCO3–(aq) + H2O(l) | Ksum |
Ksum = Ksp(CaCO2)/Ka(HCO3–) = 3.36×10-9 / 4.7×10-11 = 71.5.
The first thing to notice about this is that the second equilibrium is merely the Ka2(H2CO3) equation, written backwards. At around pH=8 or lower, we can approximate that it proceeds to completion, using up almost all of the carbonate ion (See the distribution diagram above). Thus, we can assume that the concentration [CO32–] is negligibly small. Hence, adding the two equations together and cancelling out the CO32– from the equation is a good approximation. If the pH were lower than, say, 5 then we could say the same for the Ka1(H2CO3) equilibrium.
From the Summed equation, we see that, according to Le Châtelier, if we add acid to the solution in equilibrium, the overall equilibrium will shift right to use up some of the extra acid. This will cause the solubility of the CaCO3 to increase.
Example: Calculate the solubility of CaCO3 in a buffered solution of pH=8.[1]
Since the second equilibrium (above) goes to completion, we can use the summed equilibrium.
| CaCO3(s) + | H3O+(aq) | ⇄ | Ca2+(aq) | + HCO3–(aq) | + H2O(l) | |
| I | 1×10-8 | 0 | 0 | |||
| C | Buffered means no change in [H3O+] | +x | +x | |||
| E | 1×10-8 | x | x | |||
![\[K_{sum}=\frac{[\mathrm{Ca}^{2+}][\mathrm{HCO}_3^+]}{[\mathm{H}_3\mathrm{O}^+]}=\frac{x^2}{1.0\times10^{-8}}=71.5\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-ace99bd36a4a88af8d63c80bedc87639_l3.png "Rendered by QuickLaTeX.com")
x = 8.5×10-4 .
Another example: In this one, we can not assume that the intermediate concentrations are negligibly small. We have to deal with both equilibria in full. This complicates the mathematics as we will see below.
What is the solubility of CaF2 in a solution buffered at:
A: pH = 5.0?
B: pH = 3.0?
C: pH = 1.0?
| (1) CaF2 ⇄ Ca2+ + 2F– | Ksp = 3.45×10-11 |
| (2) HF ⇄ H+ + F– | Ka = 6.3×10-4 |
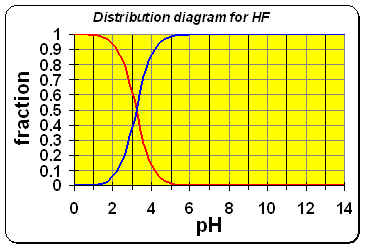
A. At pH=5, we can see from the diagram (or by comparing pKa to pH) that all the F– from the dissolution of the CaF2 remains in solution unchanged. Therefore, this particular case is just a simple Ksp question. No further calculations necessary.
| CaF2 | ⇄ | Ca2+ | + | 2F– | |
| I | 0 | 0 | |||
| C | +x | +x | |||
| E | x | x |
Ksp= 3.45×10-11 = x2 So x = 5.9×10-6
The solubility of CaF2 at pH = 5 (or higher) is 5.9×10-6 M.
B. At pH=3, we can see from the distribution diagram that some of the F– (BLUE) that came from the CaF2 will be protonated into HF (RED). The ratio (read off the diagram or calculate using the Clausius Clapeyron equation) HF to F– is approximately
.61:.39 ~ 1.56 : 1, i.e.,
[HF] = 1.56[F–].
Thus, we must deal with the two equilibria together with no assumptions possible.
Now, according to the first equilibrium, the amount of HF produced is two times the amount of CaF2 dissolved or twice the amount of Ca2+ produced.
[F]total = 2[Ca2+]
The F– that comes from the CaF2 does not all stay in that form. Some of it becomes HF. The total amount of fluorine however remains the same and can be represented by the relationship
[F]total = [HF] + [F–].
[HF] + [F–] = 2[Ca2+]
(1.56 + 1)[F–] = 2[Ca2+]
[F–] = 0.781[Ca2+].
If s is the solubility, then [Ca2+] = s.
Ksp = [Ca2+][F–]2 = s(0.781s)2 = 3.45×10-11
s = 3.8×10-4.
Obviously, at this lower pH, the solubility of the CaF2 is higher
C. At pH=1, we can assume that all the F– is now converted into HF and add equation 1 and 2 together.
| (a) | CaF2 | ⇄ | Ca2+ + 2F– | Ksp | = 3.45×10-11 |
| (b) | HF | ⇄ | H+ + F– | Ka | = 6.3×10-4 |
| (c) = a-2b | CaF2 + 2H+ | ⇄ | Ca2+ + 2HF | K |
= 3.45×10-11 × 1/(6.3×10-4)2 = 8.7×10-5 |
So now, our ICE table is made using this new final equation.
| CaF2 | + | 2H+ | ⇄ | Ca2+ | + | 2F– | |
| I | 0.1 | 0 | 0 | ||||
| C | +x | +x | |||||
| E | 0.1 | x | x |
and we now have:
![\[K=\frac{x(2x)^2}{0.1^2}=8.7\times10^{-5}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-d25c0827e291ea45a8911e2700c17c75_l3.png "Rendered by QuickLaTeX.com")
(Buffered at pH = 1 means H+ conc doesn’t change)
x = 6.0×10-3
We use these ideas of equilibrium and the application of LP to selectively alter the solubilities of ionic compounds in the qualitative analysis labs.
13.5: Metal Sulfides
There are many Industrial applications where it is important to control the solubility of metal sulfides. We can easily adjust the solubility of these ions using pH such that we can selectively precipitate Metal Sulfide solids from a mixture and effectively separate the metal ions as we filter off the precipitates in turn.
Metal II Sulfides: (Metal has oxidation state of +2; for example, M = Zn2+, Fe2+, …)
1) MS(s) ⇄ M2+ (aq) + S2-(aq)
and
2) S2–(s) + H2O(l) ⇄ HS–(aq) + OH–(aq)
However, the S2- ion is a strong base (Kb~105) and will react immediately to form HS– and a hydroxide ion. So, the two the concentration of S2- in solution is negligible and therefore, the actual dissolution process can be approximated as the sum of the reactions 1 and 2 above.
sum: MS(s) + H2O(l) ⇄ M2+ (aq) + HS–(aq) + OH–.
Thus, the true Ksp for the dissolution of a metal II sulphide is.
Ksp = [M2+][HS–][OH–]
Here we see that the addition of acid will use up OH– and hence, shift the summed equilibrium to the right thus dissolving more of the salt (MS). Since the Solubility is higher in acid solution and quite low in base solution, it is often more convenient (and conventional) to rewrite the equation for the dissolution in an acidic solution. We can use Kw = [H3O+][OH–] to do this.
MS(s) + 2H3O+(aq) ⇄ M2+ (aq) + H2S(aq) + 2H2O(l)
We call such an equilibrium constant Kspa for solubility-product constant in acid.
we can determine the solubility
![\[K_{spa} = \frac{\mathrm{[M^{2+}][H_2S]}}{[H_3O^+]^2}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-454aa381fb8d9b3294b285330fd7ee76_l3.png "Rendered by QuickLaTeX.com")
![\[s = \mathrm{[M^{2+}]} = \mathrm{[H_2S]}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-3009436f5d49a653bcc678929daa0ed1_l3.png "Rendered by QuickLaTeX.com")
![\[K_{spa} = \frac{s^2}{\mathrm{[H_3O^+]^2}}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-a74f569d52618de8fd4f9d8644ced877_l3.png "Rendered by QuickLaTeX.com")
![\[s = \sqrt{K_{spa}}\times \mathrm{[H_3O^+]}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-5a7e3ca567d1064cf7eeb151e0618a63_l3.png "Rendered by QuickLaTeX.com")
We can see that as the pH is lowered (higher H3O+ concentration) the solubility of the metal sulfide increases.
For example:
| pH = 3 | pH = 1 | ||
| Kspa(FeS) = 6×102 | ==> | s = 0.024 | s = 2.4 |
| Kspa(ZnS;Wurtzite) = 3×10-2 | ==> | s = 1.7×10-4 | s = 1.7×10-2 |
If we add acid slowly, the FeS will dissolve first (pH = ca.3-4) since it’s solubility is larger for a given [H+]. As we continue to add acid, the ZnS will eventually dissolve as well
(pH => -1).
If we had a solution containing Zn2+ and Fe2+ (both at 0.10 M) we could selectively precipitate the Zn by buffering the pH to 2.38. At that pH, the solubility of FeS is exactly 0.1 while that of the ZnS is 7.0×10-4.
So, assuming [H2S] = 0.1M, at that pH, the ZnS would be mostly in the solid form but the FeS would be Just soluble. If the pH were to raise above 2.38 or if the amount of H2S were to be increased by even the tiniest amount, then some FeS would start to form ppt as well.
There are many methods of adding reagents to a mixture of ions to selectively separate out the individual components. Various types of reactions are covered in the Qualitative Analysis lab.
NOTE: in this video, at about 36 seconds, I say “until one of them starts dissolving”. That’s a miss-spoken word. I should have said “until one of them starts precipitating.”
EXAMPLE: You have a sample containing both Iron and Zinc ions, both at a concentration of 0.10 M. The initial pH of the solution is 0, i.e., the [H+] = 1.0 M. To what pH must you change the solution to get maximum separation of the iron and zinc ions if the H2S nominal concentration is also 0.10 M?
First, the words “maximum separation” means we want to precipitate one of the ions as a salt while leaving the other ion in solution. Since it is impossible to completely separate the ions, we look for the conditions that give us the best outcome possible. This will occur when one of the ions is just barely in solution (at Ksp but with no solid yet formed), while the other is already mostly precipated.
We will be likely working in the low pH range since at high pH, the solubilities of both ions with sulfide is increased.
Hence, we’ll use the Kspa. setup. since the concentrations of the ions are fixed by the experimental conditions, we need only specify the equilibrium conditions as having a single unknown, [H3O+] = x.
| MS(s) + | 2 H3O+(aq) | ⇄ | M2+ (aq) | + H2S(aq) | + 2H2O(l) | |
| E | x | 0.10 | 0.10 |
Using the defined equilibrium values, we can substitute into our Kspa equation.
![\[K_{spa}=\frac{[\mathrm{M}^{+2}][\mathrm{H}_2 \mathrm{S}]}{\mathrm{[H_3O^+]^2}}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-57dd5cd75f34a867947a86a4d4d1b9b5_l3.png "Rendered by QuickLaTeX.com")
for FeS, we get
![\[6\times 10^2=\frac{[0.10][0.10]}{x^2}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-4fc5e16d809e5fde982e33cf67582fc2_l3.png "Rendered by QuickLaTeX.com")
![\[x=4.1\times 10^{-3}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-85df94ee4952982b1ab6ccc24a8faa24_l3.png "Rendered by QuickLaTeX.com")
for ZnS, we get
![\[3\times 10^{-2}=\frac{[0.10][0.10]}{x^2}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-f4dd1696f406669f6cdf2896f647b50d_l3.png "Rendered by QuickLaTeX.com")
![\[x=0.58\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-972cf92292f83d952e06c64be1cd2484_l3.png "Rendered by QuickLaTeX.com")
To precipitate either or both of these ions, we need to lower the [H+] by slowly adding a base, for example, NaOH. Clearly, the Fe2+ will precipitate at a lower concentration of H+ than will the Zn2+. So as we lower the concentration of the acid through 0.58, The Zn2+ will begin to precipitate. When we reach a value of [H+] = 4.1×10-3 most of the Zn2+ will have already precipitated and none of the Fe2+. At this point, one more drop of base would make the FeS start to precipitate so we don’t go there. We have reached the point of maximum separation.
13.6: Complex Ion Equilibria
Definition:
- Complex Ion:
- An ion consisting of a metal ion surrounded by ligands.
- Ligand:
- A molecule or ion having a lone-pair of electrons which can be ‘donated’ to a metal ion to form a covalent bond.
Some common ligands include H2O, NH3, Cl–, CN–.
- Coordination Number
- The number of ligands attached to a central cation.
Metal ions in Solution form complexes by covalently bonding to some number of ligands. The bond is a special kind of bond where the ligand donates one or more of it’s lone-pairs of electrons to one of the empty orbitals in the d-shell of the metal ion. These interactions can almost always be written as an equilibrium with all the requisite properties of equilibrium being valid. The reactions are always written so that one mole of the complex is the only product. Thus, these particular equilibrium constants are called formation constants or stability constants. for example, if we mix silver ions with ammonia we can observe the following two complexation reactions.
| 1) Ag+(aq) + NH3(aq) ⇄ Ag(NH3)+(aq) | Kcx1 = 2.1×103. |
| 2) Ag(NH3)+(aq) + NH3(aq) ⇄ Ag(NH3)2+(aq) | Kcx2 = 8.2×103. |
Where Kcx1 and Kcx2 are the formation constants for the two complexes Ag(NH3)+(aq) and Ag(NH3)2+(aq), respectively.
If a large enough excess of NH3 is present we can consider that the two reactions essentially go to completion and hence, the intermediate concentrations of Ag(NH3)+(aq) are very small (We check this later in the calculation). The overall reaction would then be
Ag+(aq) + 2 NH3(aq) ⇄ Ag(NH3)2+(aq)
where
Kf = Kcx12 = Kcx1×Kcx2 = 1.7×107
NOTE: If we add two reactions together as above, their equilibrium constants can be multiplied to determine the overall equilibrium constant.
These formation constants are commonly given a special symbol Kn or βn (n = coordination #) to represent the fact that the equilibrium is the formation of n-coordinate complexes. Thus, Kcx12 could also be called β2 since it represents the complexation of two ligand ammonia molecules on a silver cation.
Personally, I prefer not to use the βn notation since it can be too easily confused with other symbols. The subscript cx is my own notation just to remind myself that these are complexation reactions.
What would happen in a solution prepared by mixing 100.0 mL of 2.0 M NH3 with 100.0 mL of 1.0×10-3 M AgNO3. Let’s for a minute consider the species which will be present in the solution.
Ag+, NO3–, NH3, and of course H2O.
Possible reactions are the two complexations mentioned above and the acid-base interaction of the ammonia with water
NH3(aq) + H2O(l) ⇄ NH4(aq) + OH–(aq) Kb = 1.8×10-5.
The extent of reaction of this equilibrium is very insignificant when compared to the complexation reaction so we will ignore it. Hence, the only chemical system of interest is the complexation equilibria 1) and 2) above.
Since there is an excess of ammonia and the K is large, we can start with the assumption of 100% reaction (for proof, see footnote [2]) and then work backwards through the individual reactions to determine the actual intermediate concentrations [Ag+] and [Ag(NH3)+].
Starting with the second equilibrium, we can find the value for [Ag(NH3)+].
the assumption of 100% reaction allows us to set
[Ag(NH3)2+] = 1.0×10-3 ÷ 2 = 5.0×10-4 M.
| Ag+(aq) | + | NH3(aq) | ⇄ | Ag(NH3)+(aq) | |
| I | 0 | 1.0 | 5.0×10-4 | ||
| C | +x | +x | -x | ||
| E | x | 1.0+x | 5.0×10-4-x |
We now assume that x is small c.f. 5.0×10-4, which means, it’s also small compared to the ammonia concentration,
so we replace 1.0+x by 1.0 and 5.0×10-4–x by 5.0×10-4.
![\[K_{cx2}=\frac{[\mathrm{Ag}(\mathrm{NH}_3)_2^+]}{[\mathrm{Ag}(\mathrm{NH}_3)^+][\mathrm{NH}_3]} =8.2\times10^3=\frac{5.0\times10^{-4}}{x\times 1.0}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-5ad97dce846843f088945c63c18634d8_l3.png "Rendered by QuickLaTeX.com")
x = 6.1×10-8 so [Ag(NH3)+] = 6.1×10-8 M
check assumption: 6.1×10-8 << 5.0×10-4 (good)
Now we can use this value of [Ag(NH3)+] (x) to calculate [Ag+] using the first equilibrium.
| Ag+(aq) | + | NH3(aq) | ⇄ | Ag(NH3)+(aq) | |
| I | 0 | 1.0 | 6.1×10-8 | ||
| C | +x | +x | -x | ||
| E | x | 1.0+x | 6.1×10-8 -x |
we assume that x is small here to simplify the calculation, i.e., 1.0+x = 1.0
![\[K_{cx2}=\frac{[\mathrm{Ag}(\mathrm{NH}_3)_2^+]}{[\mathrm{Ag}(\mathrm{NH}_3)^+][\mathrm{NH}_3]} =2.1\times10^3=\frac{6.1\times10^{-8}}{x\times 1.0}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-6438e012e963c584713b4cd411a37ed5_l3.png "Rendered by QuickLaTeX.com")
x = 2.9×10-11 M = [Ag+]
check assumption: 2.9×10-11 << 6.1×10-8 (good).
Complex Ion equilibrium calculations can be relatively simple if the ligand is in large enough excess, even though the whole process looks a bit messy at first.
13.6.1: Complex Ions and Solubility
Because we can use complexation reaction reactions to ‘tie up’ metal ions in water, we can use these to increase the solubility of metal ion salts. For example, silver chloride is weakly soluble in water but quite readily dissolves in concentrated ammonia.
| AgCl(s) | ⇄ Ag+(aq) + Cl–(aq) | Ksp = 1.6×10-10 |
| Ag+(aq) + NH3(aq) | ⇄ Ag(NH3)+(aq) | Kcx1 = 2.1×103 |
| Ag+(NH3)(aq) + NH3(aq) | ⇄ Ag(NH3)2+(aq) | Kcx2 = 8.2×103 |
In this case, we cannot increase the solubility by adding acid as we did in previous examples because Cl– is a very weak base (‘very weak base’ means ‘not a base’ for our purposes). In fact, since the ammonia that is complexing with the Ag+ is a base, we will decrease the solubility by adding acid because the acid will use up some of the ammonia, thereby releasing the silver ions tied up in the complex.
If we consider there to be an excess of ammonia then we can assume these three reactions to be going essentially to completion. Thus, write an overall reaction which is the sum of the three equilibria,
| AgCl(s) + 2 NH3(aq) | ⇄ Ag(NH3)2+(aq) + Cl–(aq) |
K for this reaction is the product of the three equilibrium constants
K = KspKcx1Kcx2 = 1.6×10-10 × 2.1×103 × 8.2×103 = 2.8×10-3.
Let’s calculate the solubility of AgCl in (say) using this K constant in an amonia solution, in this example, a 10.0 M ammonia solution.
| AgCl(s) + | 2 NH3(aq) | ⇄ | Ag(NH3)2+(aq) | + | Cl–(aq) | |
| I | 10.0 | 0 | 0 | |||
| C | -2x | +x | +x | |||
| E | 10.0-2x | x | x |
![\[K_{cx2}=\frac{[\mathrm{Ag}(\mathrm{NH}_3)_2^+][\mathrm{Cl}^-]}{[\mathrm{NH}_3]^2} =2.8\times10^{-3}=\frac{x\times x}{(10.0-2x)^2}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-1c90e103bc8e141b3a9321ef91dd2159_l3.png "Rendered by QuickLaTeX.com")
Take the square root of both sides:
![\[\sqrt{2.8\times10^{-3}}=\frac{x}{10.0-2x}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-1fea8394ff03305b08fa0fa65f896b89_l3.png "Rendered by QuickLaTeX.com")
x = 0.48 M. (x is the solubility) in 10 molar ammonia.
In pure water we can quickly calculate the solubility using the original Ksp = 1.6×10-10 .
| AgCl(s) + | ⇄ | Ag+(aq) | + | Cl–(aq) | |
| I | 0 | 0 | |||
| C | +x | +x | |||
| E | x | x |
Ksp = x2 ===> x = (1.6×10-10)1/2 = 1.3×10-5 …
This is a lot smaller solubility than in the NH3 because, of course, the silver chloride is ‘pulled’ into solution by the complexing action of the ammonia on the silver ions.
- Note a typo in the video 5 above. In this example, the video asks to calculate the pH of CaCO3 but it should say solubility. ↵
-
We work out the ICE table in detail to prove while the reaction essentially goes to completion, Completion does not mean literally every last reactant is gone, only that the amount of leftover is immeasurably small.
Ag+(aq) + 2 NH3(aq) ⇄ Ag(NH3)2+ I 5.0×10-4 1.0 0 C -x -2x +x E 5.0×10-4-x 1.0-2x x
We assume that x is small compared to 1.0. Thus, we simplify to the following:![\[K_{c12}=1.7\times 10^7 =\frac{[\mathrm{Ag}(\mathrm{NH}_3)_2^+]}{[\mathrm{Ag}^+][\mathrm{NH}_3]} = \frac{x}{(5.0\times 10^{-4}-x)\times(1.0-2x)^2} \]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-014e5a7637e43da24d3de84a6df0e154_l3.png "Rendered by QuickLaTeX.com")
![\[K_{c12}=1.7\times 10^7 = \frac{x}{(5.0\times 10^{-4}-x)}\]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-faaf477e4414eb57f06fa04d3ac205cd_l3.png "Rendered by QuickLaTeX.com")
At this point we look at the left-hand side and realize that 1 << 1.7×107, so we replace (1 + 1.7×107) with just 1.7×107. This whole thing reduces to = 5.0×10-4. Since the amount of change, x, is exactly the same as the amount of initial [Ag+], We would conclude that there is exactly a 100% reaction (no silver ions left over). To 2 sig. fig., that is the best we can do. This MUST be incorrect since there is a measured equilibrium constant that is not infinity (some silver ions left over). Hence, this method is not the best way to solve this particular equilibrium problem. ↵![\[ 8.5\times10^3 = (1 + 1.7\times 10^7)x \]](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-ba613cef1ee85f80a2fda6e20135a52d_l3.png "Rendered by QuickLaTeX.com")
