68 7.4 Markers of Alzheimer’s Disease and the Effect of Neurotropin
Learning Objectives
After reading this section, you will be able to:
- Discuss the novel effects of Neurotropin in treating the APP/PS1 transgenic mouse model
- Understand the experimental disease markers that were used to follow the effects of Neurotropin
- Identify which research techniques were used to measure the changes for each disease marker
Cognition
Progressive cognitive decline of varying extent characterized by features such as memory loss, impaired thinking and learning ability is the main phenotypic feature and symptom of Alzheimer’s disease. The efficacy of Neurotropin in alleviating the cognitive impairment in AD subjects was tested by the Morris Water Maze test used as a measure for spatial learning and memory function.
The NTP-treated APP/PS1 mice demonstrated a progressive decline in escape latencies and swimming length in the course of five-day training, as well as tended to concentrate in the target area of the pool during the probe test. The test results of the APP/PS1 mice administered with NTP were comparable with those of the wild-type mice, while the control APP/PS1 mice exhibited an impaired learning ability.

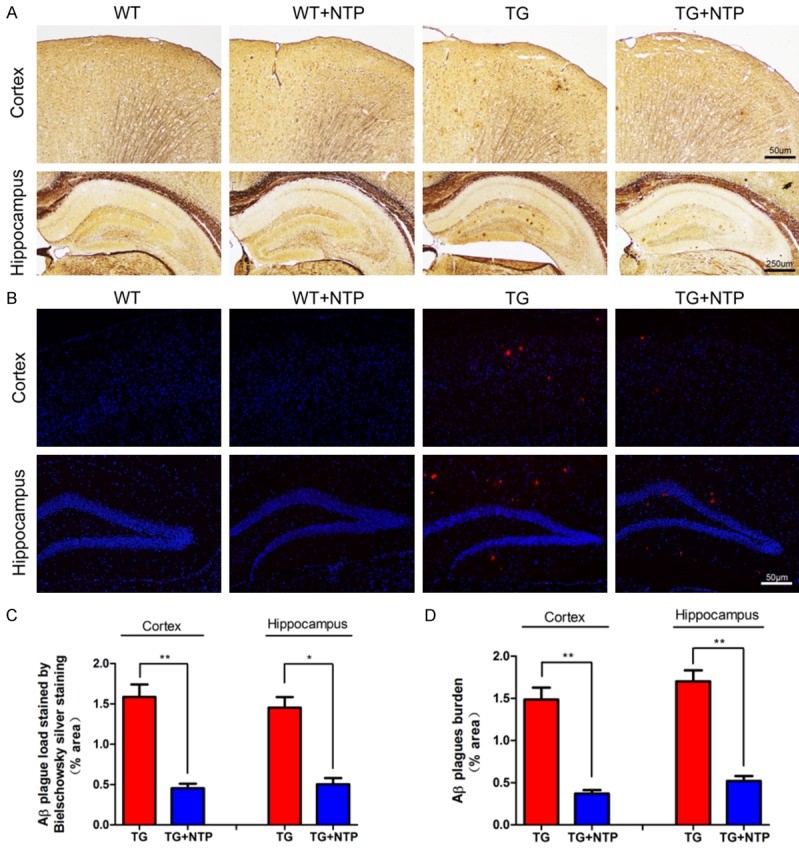
Amyloid-beta accumulation
The extracellular accumulation of amyloid-beta (Aꞵ) protein in the brain regions is one of the major characteristics of Alzheimer’s disease biology. While the direct correlation between the amount of Aꞵ plaques in the brain and the stage of dementia is weak, the extracellular amyloid-beta has been proposed to play a pivotal role in AD pathology by triggering various cascading processes such as activation of astrocytes and microglia, and subsequent neuroinflammation.
Staining the slices of hyppocampi and cortices of the four mice groups with Bielschowsky silver staining and immunofluorescent staining has revealed a significant reduction in the amyloid-beta plaques in both areas of the brain in the NTP-treated APP/PS1 mice versus the control APP/PS1 mice group.
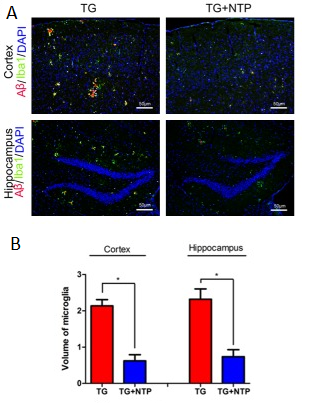
Glial Activation
Another important feature of Alzheimer’s disease, in addition to the amyloid-beta accumulation and plaque formation, is persistent neuroinflammation, which is characterized, among others, by the presence of activated microglia. The persistent activation of microglia, much like the Aꞵ accumulation, leads to various factors worsening the overall condition of AD patients. The glial activation in AD results in the production of pro-inflammatory cytokines and causes chronic neuroinflammation, pruning dysfunction and synaptic loss.
The immunofluorescent staining has revealed that amyloid-beta plaques in the hippocampi and cortices of the control APP/PS1 mice were surrounded by activated microglial cells. It was discovered that in the NTP-treated APP/PS1 mice’ brains the area occupied by the microglia was significantly reduced when compared to the APP/PS1 control group.
Cytokine Expression
In addition to examining the changes in glial cell activation, changes in the levels of pro-inflammatory cytokines were subsequently observed. The continued activation of microglia and astrocytes can lead to an increase in pro-inflammatory cytokines which at chronically elevated levels are associated with inflammatory neuronal damage and Aβ deposition (Calsolaro and Edison, 2016). The researchers used ELISA to measure the concentrations of IL-1β, IL-6, and TNF-α from the hippocampus and cortex of the WT and TG mice before and after NTP treatment.
They found that NTP significantly reduced the levels of these pro-inflammatory cytokines in APP/PS1 mice, whereas no changes were seen when WT mice were treated (figure 8.5.4). This finding reinforces the significance of NTP’s effects on CNS glial cells, demonstrating the potential for NTP to be used in managing the inflammatory reaction that causes neuronal damage in APP/PS1 mice, as well as preventing the deposition of Aβ caused by neuroinflammation.

BDNF Expression
Brain-derived neurotrophic factor (BDNF) is a growth factor that is crucial in neuron survival, growth, and plasticity. Because of this, it is also important for cognitive processes such as learning and memory. BDNF has also been found to have the ability to reduce levels of pro-inflammatory cytokines, suggesting BDNF as having a regulatory function in inflammation. However, in AD brains it has been found at reduced levels postmortem and at the mild cognitive impairment (MCI) stage of AD development (Tanila, 2017). The authors used immunofluorescent staining and ELISA to measure the changes in BDNF concentrations after NTP treatment.
Before treatment, APP/PS1 mice had significantly lower amounts of BDNF in both the hippocampus and cortex compared to WT mice. After NTP treatment, BDNF levels in TG mice increased significantly, although not to the same levels of that seen in the WT groups (figure 8.5.5). This finding signifies the potential use of NTP in restoring levels of BDNF in APP/PS1 mice, possibly explaining their improved abilities in learning and memory. This also suggests that NTP indirectly regulates neuronal inflammation by improving BDNF expression. The authors further explored this potential relationship in their next experiment.

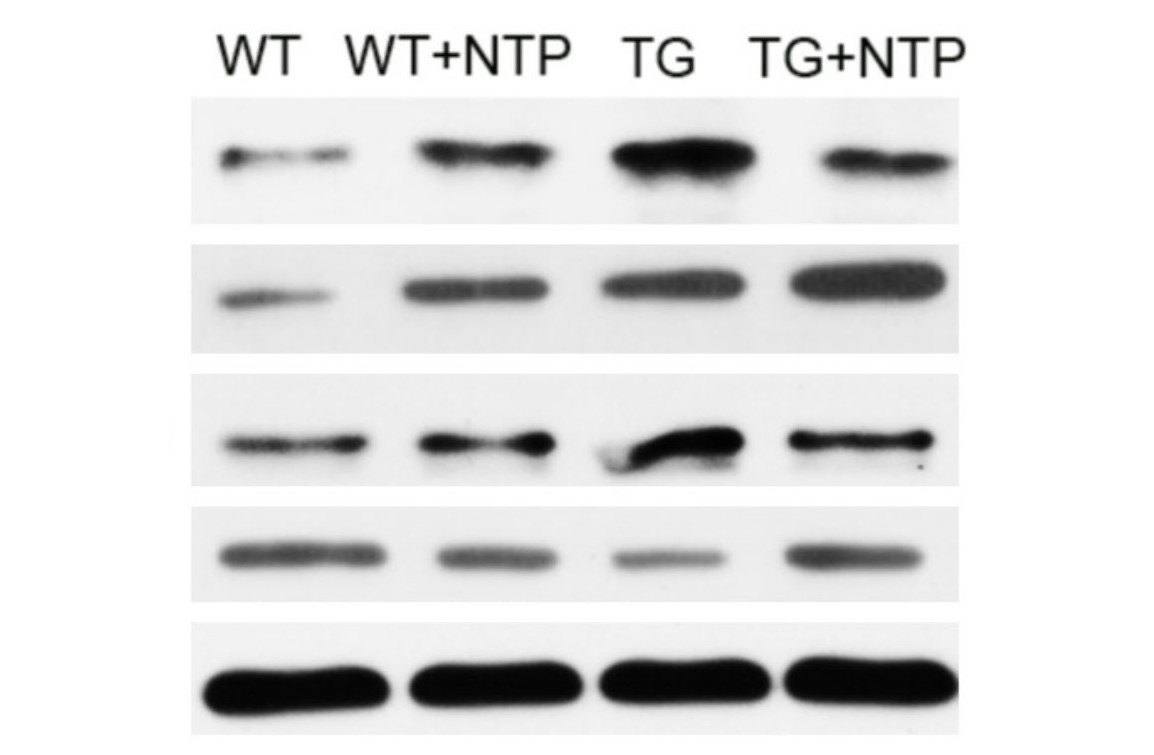
NF–κB Pathway
To potentially explain the mechanism of NTP, the authors followed the effects of NTP on the NF-κB pathway. This pathway is of interest in AD research due to its central role as a mediator of pro-inflammatory gene expression and therefore function in promoting cytokine-induced neuronal damage (Liu et al., 2017). In order to observe the changes in the NF-κB pathway, the authors used Western blot analysis to measure the changes in expression of NF-κB pathway elements, namely p-p65 and p-IκB-α.
It was found that APP/PS1 mice had elevated levels compared to WT mice, as expected. After NTP treatment, the TG mice were found to have significantly lower levels of these proteins. This finding would suggest the potential mechanism by which NTP functions to reduce neuroinflammation (figure 8.5.6). By enhancing the expression of BDNF which has a regulatory role in reducing inflammation, BDNF then acts on the NF-κB pathway to reduce the transcription and therefore expression of pro-inflammatory cytokines.
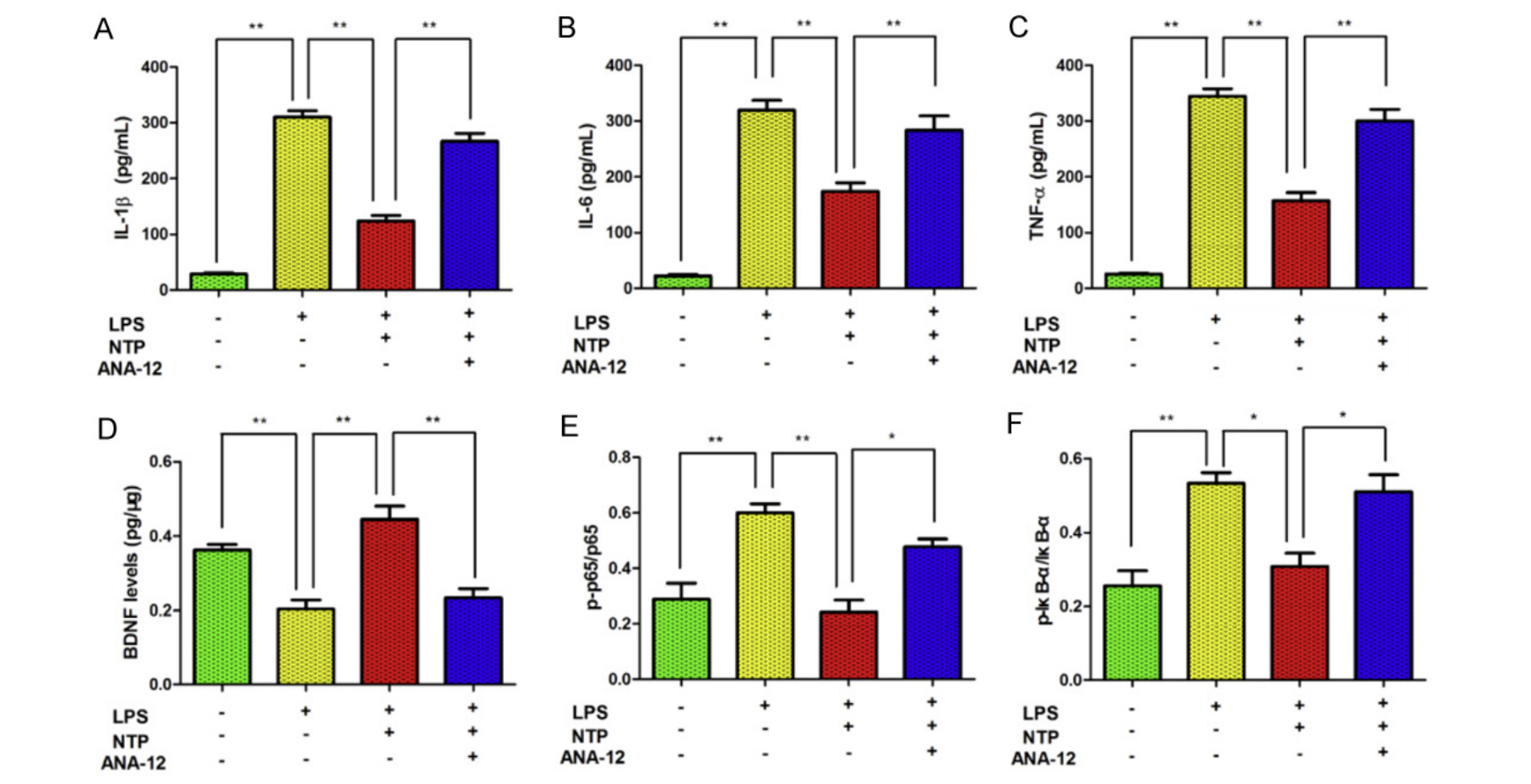
NTP Regulates the NF-κB Pathway in vitro
The authors then turned to in vitro experiments to verify and further explore the interaction between NTP and the BDNF/NF-κB pathway. These experiments involved mimicking the inflammatory reaction seen in APP/PS1 mice by treating BV-2 cells with LPS. Researchers also used a non-competitive BDNF receptor antagonist called ANA12 to observe what would happen if the proposed target molecule of NTP was inhibited.
With induced inflammation, pro-inflammatory cytokine and NF-κB pathway element levels increased, while BDNF levels decreased. After treatment with NTP, BDNF levels were increased while pro-inflammatory cytokine and NF-κB pathway element levels decreased, replicating the effects seen in vivo. When treated with ANA12, the effects of NTP were abolished, reverting BDNF, pro-inflammatory cytokine, and NF-KB pathway element levels to pre-NTP treatment levels (figure 8.5.7). This further suggests NTP’s mechanism of action via an interaction with the BDNF/NF-κB pathway.
Key Takeaways
- NTP improved the memory and learning ability of the APP/PS1 mice
- NTP significantly reduced amyloid-beta plaques and surrounding activated microglia in APP/PS1 mice in both cortex and hippocampus
- NTP was found to reduce the levels of pro-inflammatory cytokines including IL-1β, IL-6, and TNF-α in APP/PS1 mice
- BDNF, which is a crucial survival factor for neurons, was found to have enhanced expression after treatment with NTP in the APP/PS1 mice, suggesting a potential mechanism of action for NTP
- Protein elements of the NF-κB pathway were found to be decreased both in vivo and in vitro after NTP treatment in transgenic mice and BV-2 glial cells
- The proposed mechanism of action of NTP was suggested to be through an interaction with the BDNF/NF-κB pathway and was explored with the use of ANA12, finding that the inhibition of BDNF abolishes the effects of NTP
References:
Mild Cognitive Impairment (MCI) is a condition that involves the loss of memory and cognition at a rate greater than normal aging would cause, potentially indicating the progression towards dementia. In the study of AD, MCI is considered to have the early neuropsychological features of AD and therefore can be indicative of those at risk for AD (Bondi et al., 2017; Angelucci et al., 2010).
p65 is one of five components that make up the NF-κB transcription factor. The p-p65 form of the subunit denotes phosphorylation and therefore activation of the transcription factor (Giridharan and Srinivasan, 2018).
p-IκB-α is the phosphorylated and therefore activated form of IκB-α. IκB-α is one of several other IκB proteins that form a complex that inhibits the translocation of NF-κB into the cell nucleus. When phosphorylated, the IκB complex releases from NF-κB, allowing it move into the nucleus to perform its function (Giridharan and Srinivasan, 2018).
BV-2 cells are an immortalised microglial cell line derived from murine mice. They are used to primarily model microglial cells in vitro and are known to have a similar inflammatory response to that seen in vivo (Henn et al., 2009)
Lipopolysaccharide (LPS) is an endotoxin found on the outermost surface of the outer membrane of most gram negative bacteria. LPS is known to cause a potent inflammatory reaction in most mammals and is therefore commonly used in experiments as an inflammatory stimulus (Fritsche et al., 2015).