14.2 Structure et séquençage de l’ADN
Objectifs d’apprentissage
À la fin de cette section, vous serez en mesure de faire ce qui suit :
- Décrire la structure de l’ADN
- Expliquer la méthode de séquençage de l’ADN de Sanger
- Discuter des similitudes et des différences entre l’ADN eucaryote et procaryote
Les éléments constitutifs de l’ADN sont les nucléotides. Les composants importants du nucléotide sont une base azotée (contenant de l’azote), un sucre à 5 atomes de carbone (pentose) et un groupe phosphate (Figure 14.5). Le nucléotide est nommé en fonction de la base azotée. La base azotée peut être une purine telle que l’adénine (A) et la guanine (G), ou une pyrimidine telle que la cytosine (C) et la thymine (T).
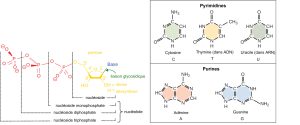
Les purines ont une structure à double cycle avec un cycle à six chaînons fusionné à un cycle à cinq chaînons. Les pyrimidines sont de plus petite taille ; elles ont une structure cyclique unique à six chaînons.
Le sucre est le désoxyribose dans l’ADN et le ribose dans l’ARN. Les atomes de carbone du sucre à cinq atomes de carbone sont numérotés 1′, 2′, 3′, 4′ et 5′ (1′ est lu « un prime »). Le phosphate, qui rend l’ADN et l’ARN acides, est relié au carbone 5′ du sucre par la formation d’une liaison ester entre l’acide phosphorique et le groupe 5′-OH (un ester est un acide + un alcool). Dans les nucléotides de l’ADN, le carbone 3′ du sucre désoxyribose est attaché à un groupe hydroxyle (OH). Dans les nucléotides d’ARN, le carbone 2′ du sucre ribose contient également un groupe hydroxyle. La base est attachée au carbone 1′ du sucre.
Les nucléotides se combinent les uns avec les autres pour produire des liaisons phosphodiester. Le résidu phosphate attaché au carbone 5′ du sucre d’un nucléotide forme une deuxième liaison ester avec le groupe hydroxyle du carbone 3′ du sucre du nucléotide suivant, formant ainsi une liaison phosphodiester 5′-3′. Dans un polynucléotide, une extrémité de la chaîne a un phosphate 5′ libre et l’autre extrémité a un 3′-OH libre. On les appelle les extrémités 5′ et 3′ de la chaîne.
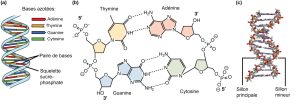
Dans les années 1950, Francis Crick et James Watson ont travaillé ensemble pour déterminer la structure de l’ADN à l’Université de Cambridge, en Angleterre. D’autres scientifiques comme Linus Pauling et Maurice Wilkins exploraient également activement ce domaine. Pauling avait précédemment découvert la structure secondaire des protéines en utilisant la cristallographie aux rayons X. Dans le laboratoire de Wilkins, la chercheuse Rosalind Franklin utilisait des méthodes de diffraction des rayons X pour comprendre la structure de l’ADN. Watson et Crick ont pu reconstituer le casse-tête de la molécule d’ADN sur la base des données de Franklin parce que Crick avait également étudié la diffraction des rayons X (Figure 14.6). En 1962, James Watson, Francis Crick et Maurice Wilkins ont reçu le prix Nobel de médecine. Malheureusement, Franklin était décédé à ce moment-là et les prix Nobel ne sont pas décernés à titre posthume.

Watson et Crick ont proposé que l’ADN soit composé de deux brins qui sont enroulés l’un autour de l’autre pour former une hélice dextrogyre (qui tourne dans le sens des aiguilles d’une montre). L’appariement des bases a lieu entre une purine et une pyrimidine sur des brins opposés, de sorte que A s’apparie avec T et G s’apparie avec C (suggéré par les règles de Chargaff). Ainsi, l’adénine et la thymine sont des paires de bases complémentaires, et la cytosine et la guanine sont également des paires de bases complémentaires. Les paires de bases sont stabilisées par des liaisons hydrogène : l’adénine et la thymine forment deux liaisons hydrogène et la cytosine et la guanine forment trois liaisons hydrogène. Les deux brins sont de nature antiparallèle ; c’est-à-dire que l’extrémité 3′ d’un brin fait face à l’extrémité 5′ de l’autre brin. Le sucre et le phosphate des nucléotides forment l’épine dorsale de la structure, tandis que les bases azotées sont empilées à l’intérieur, comme les barreaux d’une échelle. Chaque paire de bases est séparée de la paire de bases suivante par une distance de 0,34 nm, et chaque tour de l’hélice mesure 3,4 nm. Par conséquent, 10 paires de bases sont présentes par tour de l’hélice. Le diamètre de la double hélice de l’ADN est de 2 nm, et il est uniforme partout. Seul l’appariement entre une purine et une pyrimidine et l’orientation antiparallèle des deux brins d’ADN peuvent expliquer le diamètre uniforme. La torsion des deux brins l’un autour de l’autre entraîne la formation de rainures majeures et mineures uniformément espacées (Figure 14.7).
Techniques de séquençage de l’ADN
Jusque dans les années 1990, le séquençage de l’ADN (lecture de la séquence d’ADN) était un processus relativement long et coûteux. L’utilisation de nucléotides radiomarqués a également aggravé le problème pour des raisons de sécurité. Avec la technologie et les machines automatisées actuellement disponibles, le processus est moins cher, plus sûr et peut être achevé en quelques heures. Fred Sanger a mis au point la méthode de séquençage utilisée pour le projet de séquençage du génome humain, qui est largement utilisée aujourd’hui (Figure 14.8).
La méthode de séquençage est connue sous le nom de méthode de terminaison de la chaîne didésoxy. La méthode est basée sur l’utilisation de terminateurs de chaîne, les didésoxynucléotides (ddNTP). Les ddNTPSs diffèrent des désoxynucléotides par l’absence d’un groupe 3′ OH libre sur le sucre à cinq atomes de carbone. Si un ddNTP est ajouté à un brin d’ADN en croissance, la chaîne ne peut pas être étendue plus loin, car le groupe 3′ OH libre nécessaire pour ajouter un autre nucléotide n’est pas disponible. En utilisant un rapport prédéterminé entre les désoxynucléotides et les didésoxynucléotides, il est possible de générer des fragments d’ADN de différentes tailles.
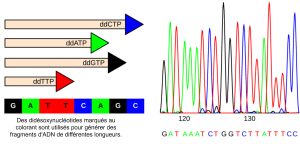
L’échantillon d’ADN à séquencer est dénaturé (séparé en deux brins en le chauffant à haute température). L’ADN est divisé en quatre tubes dans lesquels une amorce, l’ADN polymérase, et les quatre nucléosides triphosphates (A, T, G et C) sont ajoutés. De plus, des quantités limitées de l’un des quatre didésoxynucléosides triphosphates (ddCTP, ddATP, ddGTP et ddTTP) sont ajoutées à chaque tube respectivement. Les tubes sont étiquetés A, T, G et C selon le ddNTP ajouté. À des fins de détection, chacun des quatre didésoxynucléotides porte une étiquette fluorescente différente. L’allongement de la chaîne se poursuit jusqu’à ce qu’un nucléotide didésoxy fluorescent soit incorporé, après quoi aucun autre allongement n’a lieu. Une fois la réaction terminée, une électrophorèse est effectuée. Même une différence de longueur d’une seule base peut être détectée. La séquence est lue à partir d’un scanneur laser qui détecte le marqueur fluorescent de chaque fragment. Pour ses travaux sur le séquençage de l’ADN, Sanger a reçu un prix Nobel de chimie en 1980.
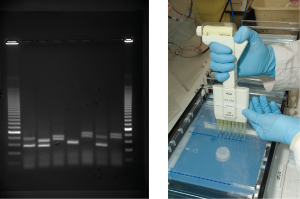
L’électrophorèse sur gel est une technique utilisée pour séparer des fragments d’ADN de différentes tailles. Habituellement, le gel est composé d’un produit chimique appelé agarose (un polymère polysaccharide extrait d’algues riche en résidus de galactose). La poudre d’agarose est ajoutée à un tampon et chauffée. Après refroidissement, la solution de gel est versée dans un plateau de coulage. Une fois le gel solidifié, l’ADN est chargé sur le gel et un courant électrique est appliqué. L’ADN a une charge négative nette et se déplace de l’électrode négative vers l’électrode positive. Le courant électrique est appliqué pendant suffisamment de temps pour permettre à l’ADN de se séparer en fonction de sa taille ; les plus petits fragments seront les plus éloignés du puits (où l’ADN a été chargé), et les fragments de poids moléculaire plus lourds seront les plus proches du puits. Une fois l’ADN séparé, le gel est coloré avec un colorant spécifique à l’ADN pour le visualiser (Figure 14.9).
LIEN AVEC L’ÉVOLUTION
Génome de Néandertal
Comment sommes-nous liés? La première ébauche de la séquence du génome de Néandertal a été récemment publiée par Richard E. Green et coll. en 2010.1 Les Néandertaliens sont les ancêtres les plus proches des humains actuels. On sait qu’ils ont vécu en Europe et en Asie occidentale (et maintenant, peut-être, en Afrique du Nord) avant de disparaître des archives fossiles il y a environ 30 000 ans. L’équipe de Green a étudié des restes fossiles vieux de près de 40 000 ans qui ont été sélectionnés sur des sites du monde entier. Des moyens extrêmement sophistiqués de préparation des échantillons et de séquençage de l’ADN ont été utilisés en raison de la nature fragile des os et de la forte contamination microbienne. Dans leur étude, les scientifiques ont pu séquencer quelque quatre milliards de paires de bases. La séquence néandertalienne a été comparée à celle des humains actuels du monde entier. Après avoir comparé les séquences, les chercheurs ont constaté que le génome de Néandertal présentait une similitude de 2 à 3 % plus grande avec les personnes vivant en dehors de l’Afrique qu’avec les personnes en Afrique. Alors que les théories actuelles suggèrent que tous les humains actuels peuvent être retracés jusqu’à une petite population ancestrale en Afrique, les données du génome de Néandertal suggèrent un certain croisement entre les Néandertaliens et les premiers humains modernes.
Green et ses collègues ont également découvert des segments d’ADN chez les personnes en Europe et en Asie qui ressemblent davantage aux séquences néandertaliennes qu’à d’autres séquences humaines contemporaines. Une autre observation intéressante est que les Néandertaliens sont aussi étroitement liés aux habitants de Papouasie-Nouvelle-Guinée qu’à ceux de Chine ou de France. C’est surprenant, car les restes fossiles de Néandertal n’ont été localisés qu’en Europe et en Asie occidentale. Très probablement, des échanges génétiques ont eu lieu entre les Néandertaliens et les humains modernes lorsque les humains modernes ont émergé d’Afrique, avant la divergence des Européens, des Asiatiques de l’Est et des Papouasie-Nouvelle-Guinéens.
Plusieurs gènes semblent avoir subi des changements par rapport aux Néandertaliens au cours de l’évolution des humains actuels. Ces gènes sont impliqués dans la structure crânienne, le métabolisme, la morphologie de la peau et le développement cognitif. L’un des gènes qui présente un intérêt particulier est RUNX2, qui est différent chez les humains modernes et les Néandertaliens. Ce gène est responsable de l’os frontal proéminent, de la cage thoracique en forme de cloche et des différences dentaires observées chez les Néandertaliens. On suppose qu’un changement évolutif dans RUNX2 a joué un rôle important dans l’origine de l’humain moderne, et cela a affecté le crâne et le haut du corps.
L’empaquetage de l’ADN dans les cellules
Les procaryotes sont beaucoup plus simples que les eucaryotes dans beaucoup de leurs caractéristiques (Figure 14.10). La plupart des procaryotes contiennent un seul chromosome circulaire qui se trouve dans une zone du cytoplasme appelée nucléotide.
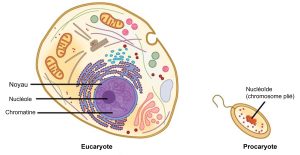
La taille du génome de l’un des procaryotes les mieux étudiés, E. coli, est de 4,6 millions de paires de bases (environ 1,1 mm, si coupé et étiré). Alors, comment cela s’intègre-t-il dans une petite cellule bactérienne? L’ADN est déformé par ce que l’on appelle le super-enroulement. Le super-enroulement suggère que l’ADN est soit « sous-enroulé » (moins d’un tour d’hélice pour 10 paires de bases), soit « superenroulé » (plus d’un tour pour 10 paires de bases) par rapport à son état détendu normal. Certaines protéines sont connues pour être impliquées dans le superenroulement ; d’autres protéines et enzymes telles que l’ADN gyrase aident à maintenir la structure superenroulée.
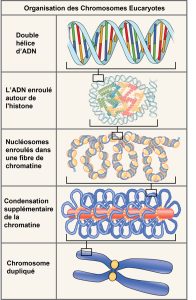
Les eucaryotes, dont les chromosomes sont chacun constitués d’une molécule d’ADN linéaire, utilisent un type différent de stratégie d’empaquetage pour adapter leur ADN à l’intérieur du noyau (Figure 14.11). Au niveau le plus élémentaire, l’ADN est enroulé autour de protéines appelées histones pour former des structures appelées nucléosomes. Les histones sont des protéines conservées au cours de l’évolution qui sont riches en acides aminés basiques et forment un octamère composé de deux molécules de chacune des quatre histones différentes. Leur composition et leurs propriétés sont importantes pour comprendre l’expression des gènes et ont été partiellement découvertes sur la base des recherches de Marie M. Daly et Alfred E. Mirsky au début des années 1950. L’ADN (rappelez-vous, il est chargé négativement à cause des groupes phosphate) est étroitement enroulé autour du noyau des histones. Ce nucléosome est lié au suivant à l’aide d’un ADN de liaison. Ceci est également connu sous le nom de structure « collier de perles». À l’aide d’une cinquième histone, une chaîne de nucléosomes est ensuite compactée en une fibre de 30 nm, soit le diamètre de la structure. Les chromosomes de métaphase sont encore plus condensés par association avec des protéines de structure. Au stade de la métaphase, les chromosomes sont les plus compacts, avec une largeur d’environ 700 nm.
En interphase, les chromosomes eucaryotes ont deux régions distinctes qui peuvent être distinguées par coloration. La région étroitement empaquetée est connue sous le nom d’hétérochromatine et la région moins dense est connue sous le nom d’euchromatine. L’hétérochromatine contient généralement des gènes qui ne sont pas exprimés et se trouve dans les régions du centromère et des télomères. L’euchromatine contient généralement des gènes qui sont transcrits, avec de l’ADN emballé autour des nucléosomes, mais pas plus compacté.
Notes de bas de page
1Richard E. Green et coll., « A Draft Sequence of the Neandertal Genome », Science 328 (2010): 710-22.
