5 Determining the Amino Acid Sequence of a Protein
Synopsis: Early methods to determine the amino acid sequence of a protein relied on cycling between basic and acidic environments to change the reactivity of the peptide. In practice, proteins need to hydrolysed into shorter peptides for sequencing. Selective hydrolysis of the polypeptide chain by proteases or with chemicals, cuts very long polypeptides into specific fragments of more manageable sizes. Using tandem mass spectrometry, proteins can be sequenced. Their identity will be determined by searching protein databases and using search tools like BLAST.
Determining amino acid sequence
Fred Sanger at Cambridge University was the first person to devise a method to determine the amino acid sequence of a polypeptide/protein. Over the period 1947-1953, he worked out methods to find the amino acid sequence of the protein hormone insulin, and eventually won the Nobel Prize for this work.
Sanger introduced two important techniques:
- N-terminal tagging identifies the first amino acid in the chain
- Limited hydrolysis breaks the chain into smaller, more manageable pieces
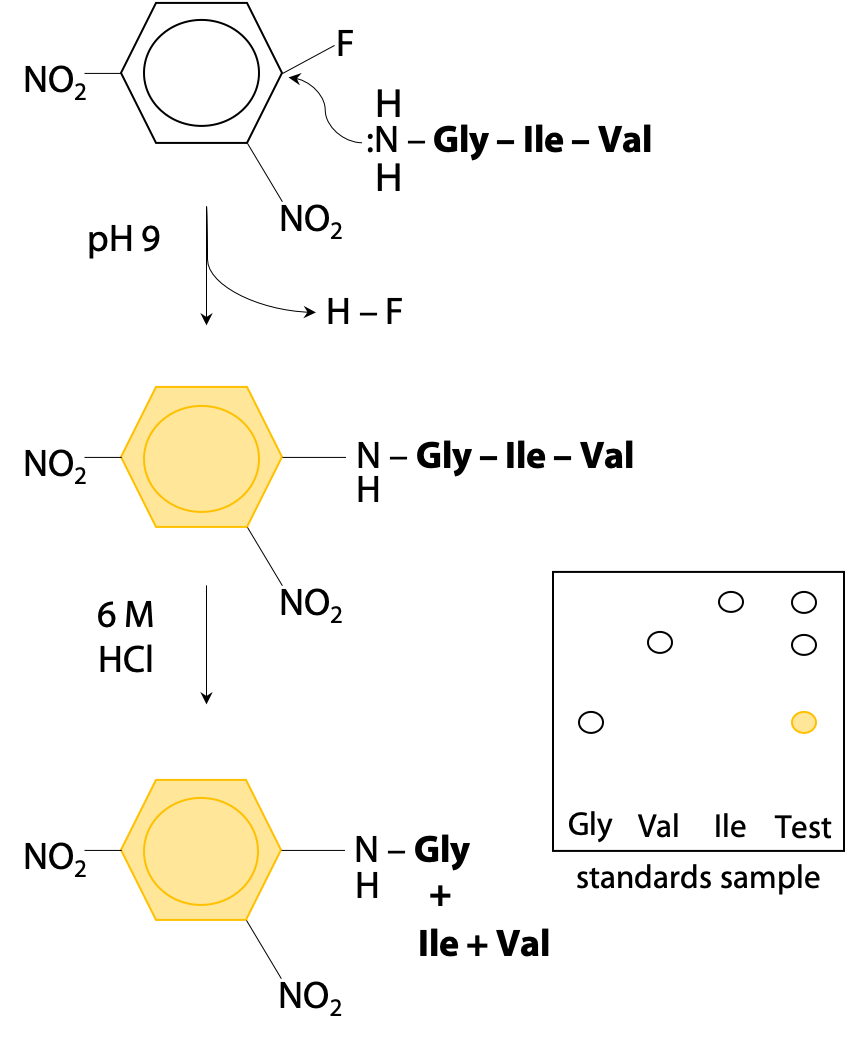
N-terminal tagging works because the N-terminal amino group becomes a nucleophile under mildly basic conditions (+NH3–, the normal state at pH 7 is not a nucleophile, but by increasing to pH 9 (pKa = 8 for the N-terminal of a peptide chain), it becomes deprotonated :NH2–.) To avoid unwanted reactions at lysine, pKa 10.2, the pH should not be raised any further.
The nucleophilic N-terminal :NH2– will then react by displacing HF from the reagent fluorodinitrobenzene (Figure 5.1).
Thus, the bright yellow dinitrophenyl group becomes bonded to the N-terminal amino acid. The tagged protein is then hydrolysed to its constituent amino acids, and the labeled (yellow) N-terminal amino acid can easily be separated and identified by chromatography.
Unfortunately, Sanger’s method requires complete hydrolysis of the peptide chain to recover the tagged amino acid. This destroys the rest of the peptide chain, so amino acids #2, #3 etc are not easily identified. Sanger proceeded by using limited hydrolysis, hydrolysis at lower temperature or for shorter time, so that not all peptide bonds are broken. This creates a random mixture of dipeptides and tripeptides (short chains of 2-3 amino acids). By analyzing all the fragments, he was able to reconstruct the whole sequence, but it took 7 years to put together all the pieces of the puzzle. Luckily for Sanger, insulin is a very small protein with two chains of 21 and 30 amino acids respectively.
Per Edman solved the problem of hydrolyzing the complete peptide to recover the tagged amino acid in Sweden in 1956. He used the reagent phenylisothiocyanate to label the N-terminal end of the polypeptide sample (Figure 5.2). (Stryer, Fig. 4.21.)
Phenylisothiocyanate reacts with a deprotonated N-terminal amino group.
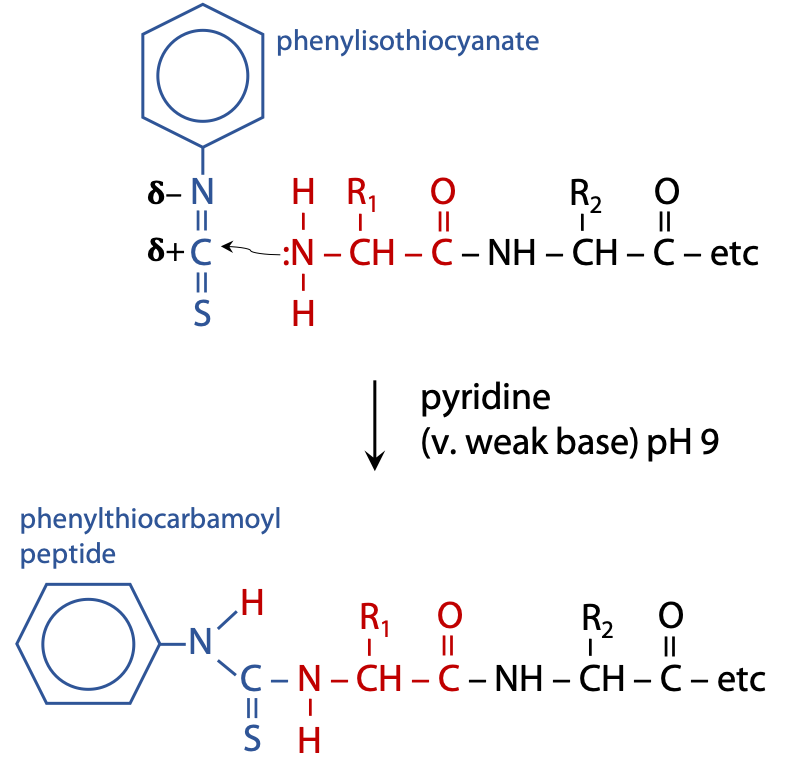
Deprotonation exposes the lone pair of N, allowing it to react as a nucleophile, which can then attack an electron deficient nucleus, the C atom of isothiocyanate. This requires mildly basic conditions, pH 9, which is achieved by carrying out the reaction in a weak base such as pyridine. The coupled product is called a phenylthiocarbamoyl peptide.
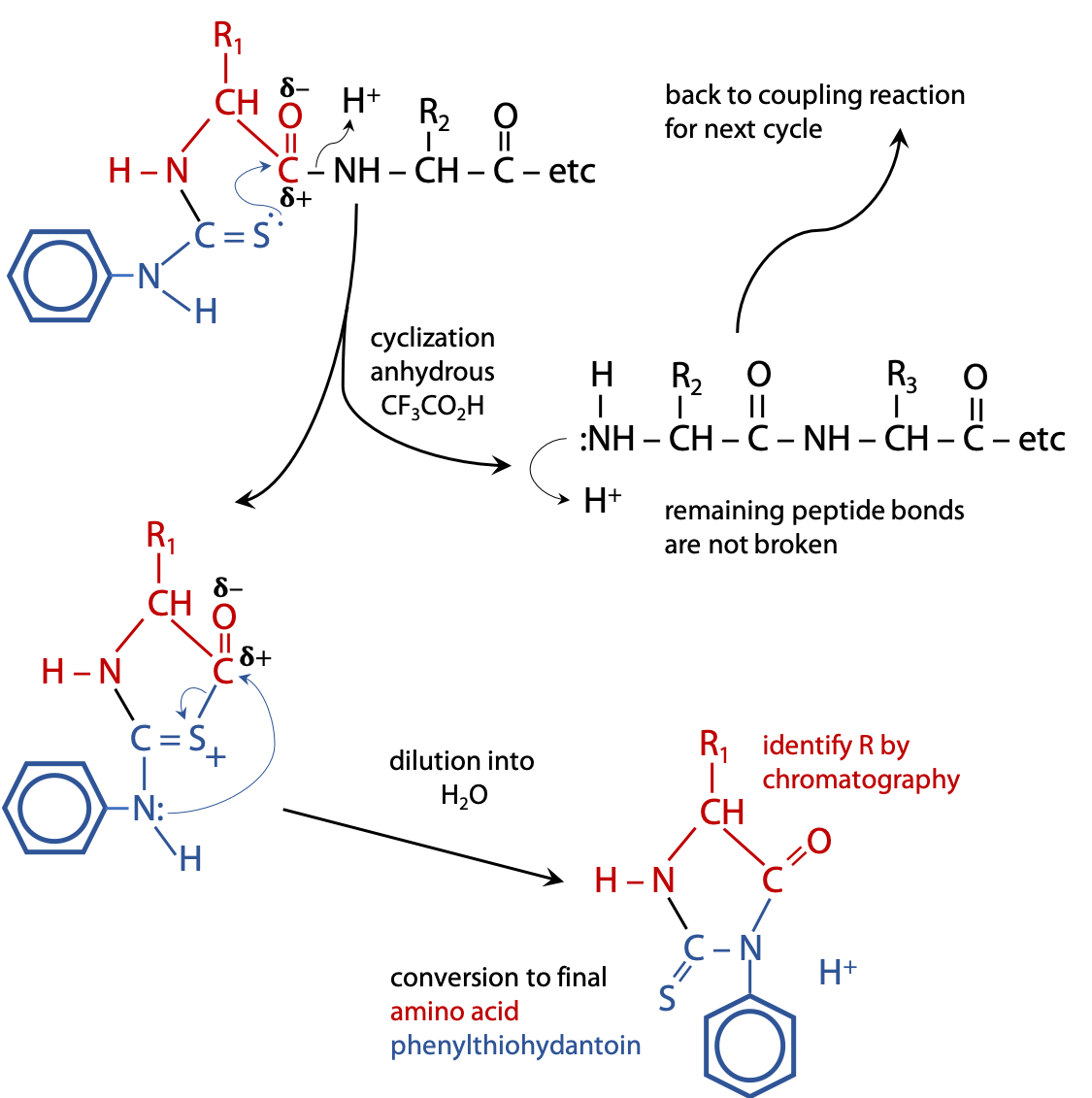
The phenylthiocarbamoyl peptide is transferred into weak anhydrous (no H2O) acid, e.g. CF3CO2H, which causes the C=S to attack the nearest peptide bond, i.e. the one linking the N-terminal amino acid to the rest of the chain. The result is a cyclization reaction that splits off the first amino acid, leaving the rest of the chain intact (Figure 5.3).
Because the process is carried out in the absence of H2O, there is no “hydrolysis“.
The cyclized form of the first amino acid rearranges to the final product, an amino acid phenylthiohydantoin or PTH amino acid.
The released amino acid phenylthiohydantoin is then identified by chromatography or mass spectrometry. Because the rest of the chain is left intact, the cycle of reactions can be repeated many times, each cycle removing the currently exposed N-terminal amino acid, allowing each to be identified in sequence:
PTH-Gly + Ile-Val-Glu-Gln-Cys-Cys-Ala-Ser-Val
PTH-Ile + Val-Glu-Gln-Cys-Cys-Ala-Ser-Val
PTH-Val + Glu-Gln-Cys-Cys-Ala-Ser-Val etc.
An important factor for success is that the two steps require contrasting conditions:
- Coupling with phenylisothiocyanate occurs in weak base
- Cyclization to phenylthiohydantoin occurs in anhydrous acid
Because there are two distinct phases to the reaction, the reaction cycle remains strictly in phase. The coupling reaction at step 1 can be allowed to go to completion without any risk that some molecules of glycine make cyclize early and expose Ile prematurely, because cyclization requires acid. Similarly at step 2, the cyclization of Gly can proceed to completion without risk of Ile coupling early, since the conditions are acidic, not basic.
Another advantage is that the cycle of reactions is very easy to automate. The whole process can be carried out by machine, producing one PTH amino acid every hour.
Although the Edman reaction can be repeated many times with high yields, there are practical limits. It’s usual to read off sequences of 20-30 amino acids in one experiment. Sequences much over 50 or 60 amino acids are very hard to handle in a single run. Even if a reaction has 98% or 99% yield, there’s a limit to the number of times you can repeat it.
To overcome this limitation, proteins are hydrolyzed into peptides that can then be sequenced.
Selective hydrolysis
Selective hydrolysis of polypeptides allows a long polypeptide to be cut at specific locations, giving shorter oligopeptides. If the oligopeptides are no longer than 20-30 amino acids, their sequences can be determined by Edman’s method or mass spectrometry.
Proteases
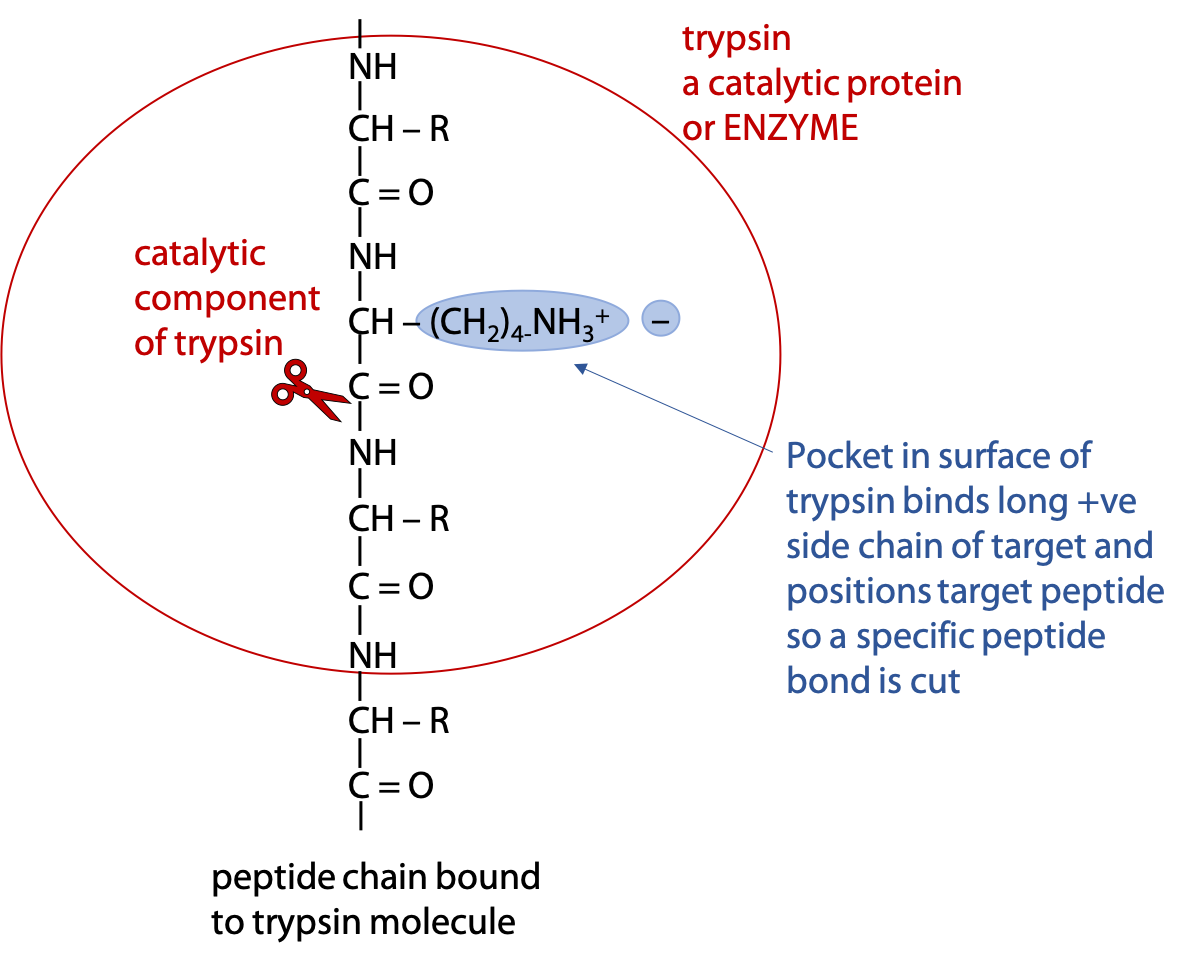
Selective hydrolysis can be achieved with the help of digestive enzymes called proteases. Enzymes are proteins that catalyze a specific reaction, which in this case, hydrolysis of the targeted peptide bond (Figure 5.4). (Stryer Section 9.1. )
Trypsin is an enzyme that binds a polypeptide and cuts the peptide bond on the carboxylate side of the targets Arg or Lys.
Chymotrypsin cuts polypeptide on the carboxylate side of Phe, Tyr or Trp. The detailed molecular mechanism of chymotrypsin is described in Chapter 9.
In both cases, if the next amino acid after the target is proline, the polypeptide fails to bind to the enzyme and can’t be cut at that point. Proline has an unusual conformation, due to the side chain bonding to the α-amino N.
e.g. for trypsin
Gly——Lys-X——-Arg-Y——Lys-Pro——Asn
2 cuts give 3 peptides
Gly——Lys + X——-Arg + Y—–Lys-Pro——Asn
and for chymotrypsin
Gly——Phe-X——-Trp-Y——Phe-Pro——Asn
2 cuts give 3 peptides
Gly——Phe + X——-Trp + Y—–Phe-Pro——Asn
Selective chemical hydrolysis
The chemical reagent cyanogen bromide, CNBr, may also be used. Cyanogen bromide attacks on the carboxylate side of methionine, converting it to homoserine, Hse. Being a chemical reagent, not a catalyst, cyanogen bromide is consumed in the reaction:
Gly—Met-X—————-Met-Y——————-Asn
2 cuts give 3 peptides
Gly—Hse + X————-Hse + Y—————–Asn
The overlap method

Different proteases would cut myoglobin at different sites, resulting in different oligopeptides that originate from the same primary sequence (Figure 5.5).
If myoglobin is digested in chymotrypsin, all the red labelled sites will be hydrolysed at the peptide bonds immediately following the target amino acid, since it’s not possible to attack at only one location at a time. Similarly all the sites labelled in blue will be cut by trypsin.
This creates a series of oligopeptides with a characteristic pattern of molar masses that is unique to a given polypeptide.
In experiments to determine the complete amino acid sequence of a protein, selective hydrolysis is first carried out, and the resulting oligopeptides are separated by chromatography. Usually ion exchange, reversed phase or gel filtration techniques are used. The individual peptides can then be sequenced by Edman’s method. Alternatively, the oligopeptide masses are easily measured by mass spectrometry (see Chapter 4) and this can be used to identify a particular protein.
After all oligopeptide sequences have been determined, the complete polypeptide sequence is deduced by the overlap method (see Stryer Section 4.2.1.)
Using mass spectrometry to sequence and identify proteins
Proteins can be sequenced directly using tandem mass spectrometry (tandem MS or MS/MS). This is a modern technique that is commonly used in the analysis of the entire protein complement of an organism or cell (an approach called proteomics). Since very small amounts of proteins are required, individual bands on 2D gels (see earlier lecture) can be cut out and sequenced without the need for complicated protein purification techniques.
The protein sample is hydrolysed into a mixture of shorter peptides using a protease or through chemical means. This mixture is then injected into a tandem MS, essentially two mass spectrometers in series (Figure 5.6).
In the first MS chamber, peptides of different masses are separated. Each of these peptides is then introduced into a collision cell where each peptide molecule fragments only once, usually at a peptide bond. In the second MS chamber, the masses of the peptide fragments are measured.
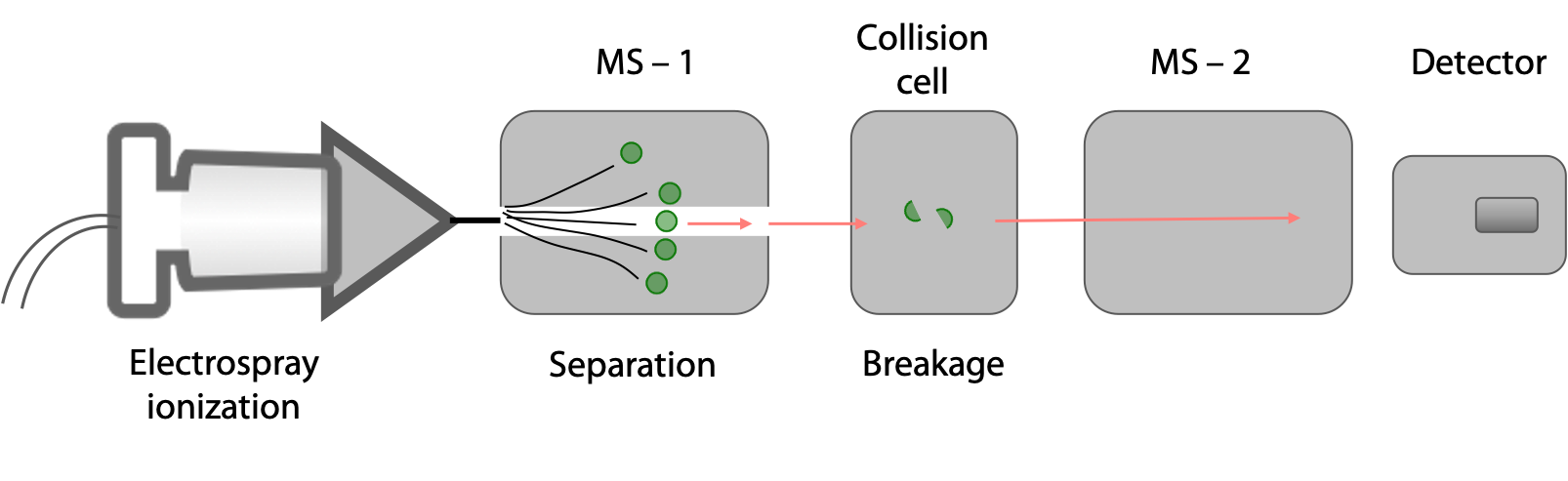
In one commonly used method, peptides are generated from the full-length protein with trypsin. Each resulting peptide must; therefore, have a Lys or Arg at its C-terminus (see page 1 for the recognition site for trypsin).
The peptide is then moved into a low pH buffer. Under these conditions, acidic residues have no charge on the sidechains, while basic residues have +1 charge on their sidechains (Figure 5.7).
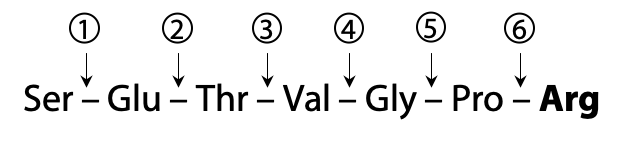
Here is an example peptide that we will use to illustrate the process of sequencing with MS:

This peptide then undergoes fragmentation, breaking one peptide bond per molecule on average, in a statistically random fashion. The example peptide might be fragmented at one of six possible break sites (Figure 5.8).
These fragments then go through the second MS, where peptides with charges produce the highest signal. The resulting mass spectra would look something like this for our example peptide (Figure 5.9):
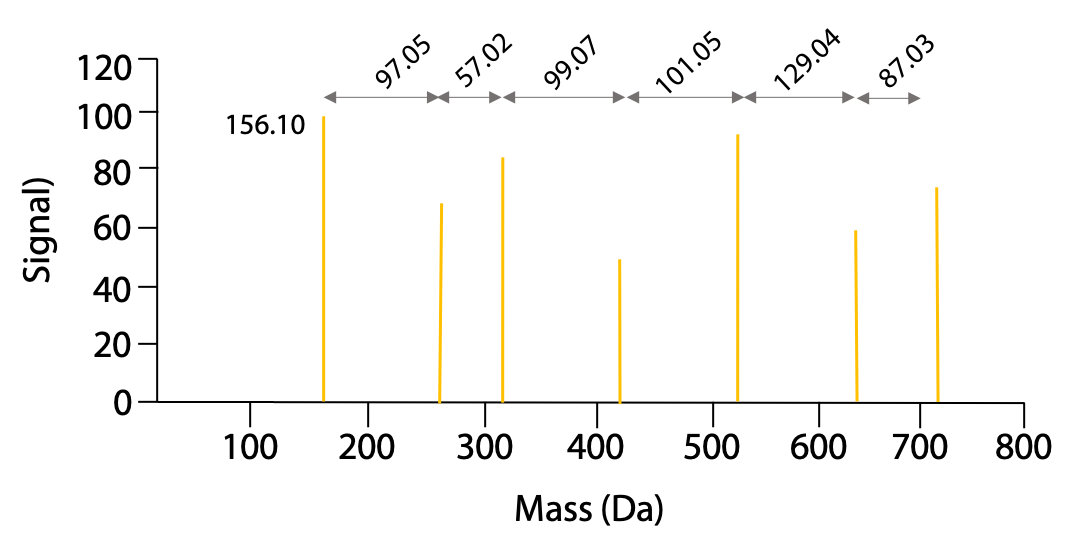
The mass of the peaks represents the mass of one charged fragment type. The difference in mass between the peaks presents the mass of one amino acid as you go from one fragment to the next. The data would be presented as follows (Table 5.1):
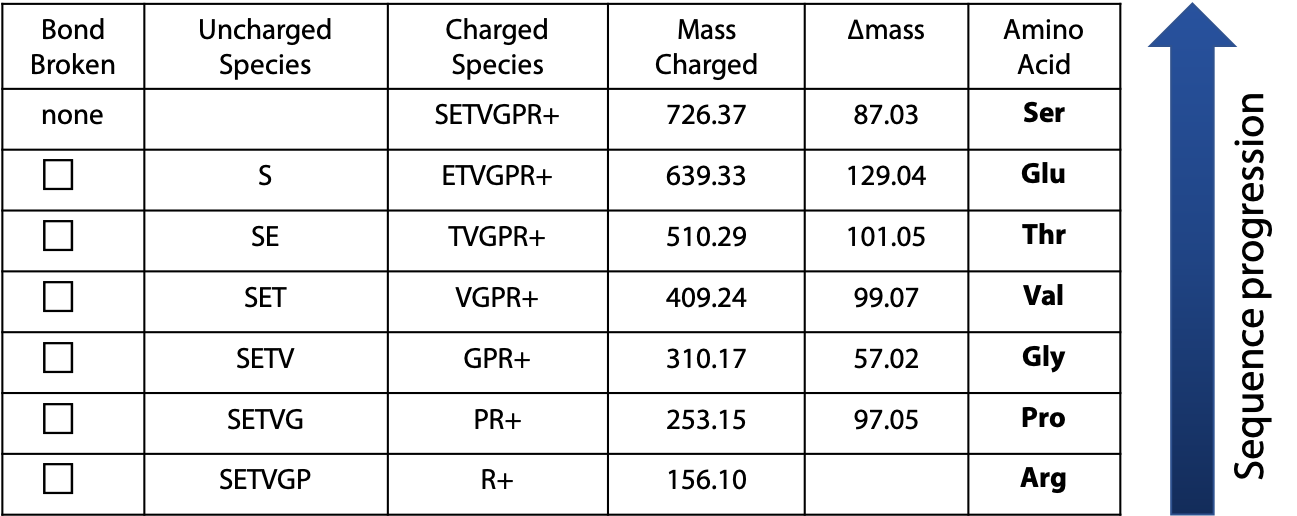
The difference in mass between fragments is used to identify the amino acid, using the following list of amino acid masses (Figure 5.10). The only ambiguity is leucine and isoleucine, which have exactly the same mass.
Amino acid residue masses (Da)

By using the progression of masses to identify the amino acids involved, the original sequence of the peptide can be assembled. In the example, the charged amino acid (Arg) was at the C-terminus. Because this amino acid served as the “anchor” for the MS due to its charge under the low pH conditions of the fragmentation, we must begin assembling the sequence from the C-terminus and progress toward the N-terminus (Figure 5.11). The resulting sequence is:

The NCBI Database and BLAST searching
The sequence of a peptide can be compared with databanks of protein sequences of all known proteins. The NCBI (National Center for Biotechnology Information) database (www.ncbi.nlm.nih.gov) contains a vast amount of sequence information. With the explosion of DNA sequencing data available today and our ability to convert those DNA sequences into the protein sequences that they encode, the amount of protein sequence information available is astronomical and still growing every day.
One tool that is available through the NCBI is a search of protein sequences, called BLAST (Basic Local Alignment Search Tool). With this tool, one can enter a peptide or protein primary sequence and generate a list of sequences that contain the highest similarity or identity.
Several results can be obtained:
- If the protein being analyzed is already in the protein database, then a 100% match will be generated.
- If it is a protein that is not in the database, but has a close relative from another organism or has a similar isoform in the same organism, then a very close match will result. These kinds of similar proteins are called homologs; e.g. myoglobin from horse and whale, α-actin and β-actin in humans. This is often enough to identify the kind of protein.
- If it is a completely new protein, then there will be very little homology in the database and the identity of the protein will not be apparent.
Beyond sequence alignments, performing biochemical tests with a purified protein is the most definitive method of determining the identity of a protein.
This technique also has the added advantage of being able to identify the location and type of any post-translational modifications that may be present on the protein. These modifications are small molecules that are covalently bound to amino acid side chains. A good example of this kind of modification is protein phosphorylation, where the mass of an added PO4 group would be added to a Ser, Thr or Tyr side chain.
Self-assessment Questions
1.
2.
3.