9 The Catalytic Mechanism of Chymotrypsin & Measuring Activity
Synopsis: Chymotrypsin catalyzes hydrolysis of a substrate peptide at the carboxylate group of phenylalanine, tyrosine or tryptophan, unless the next amino acid is proline. This behaviour depends on the coordinated action of several groups of amino acids in chymotrypsin: the binding pocket precisely positions the target amino acid; the catalytic triad provides an active nucleophile and general acid/base catalysis; the oxyanion hole stabilizes the shape of the transition state. Insight into the activity of enzymes like chymotrypsin can be obtained by measuring the rates of enzyme-catalyzed reactions through enzyme activity assays. Assays that use changes of the absorbance of a sample are simple and allow constant monitoring of reactions.
Work through the Chymotrypsin Worksheet
Read the supplementary material “What do the curly arrows mean in reaction diagrams?”
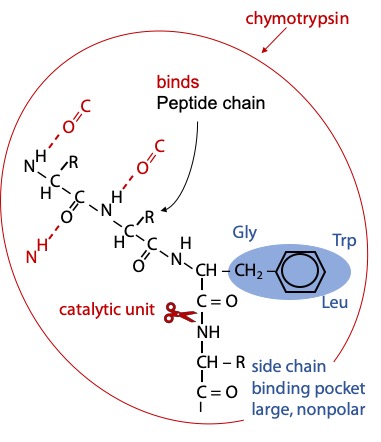
Chymotrypsin binds the substrate peptide into a groove on the surface of the enzyme. Weak binding occurs via hydrogen bonds to the NH group of the target amino acid and to several backbone CO or NH groups preceding, so the enzyme selects a peptide chain as its substrate.
Strong binding of the target amino acid occurs when its benzene ring side chain fits in a hydrophobic pocket on the surface of chymotrypsin (Figure 9.1). If the substrate peptide chain contains one of the target amino acids, the peptide bond immediately following the target is positioned precisely next to the catalytic components.
Peptide hydrolysis by H2O without the help of a catalyst

H2O acts as a nucleophile:
(1) a lone pair from the water is donated to C of C=O, due to C with a partial positive charge (bonded to electronegative O) Figure 9.2).
C can’t accommodate more than 8 valence electrons, so the electronegative O atom withdraws a bonding electron pair to itself.
The transition state forms a semistable oxyanion –O–, and the attacked C atom goes from initial sp2 state (trigonal planar) to sp3 (tetrahedral) in the transition state.
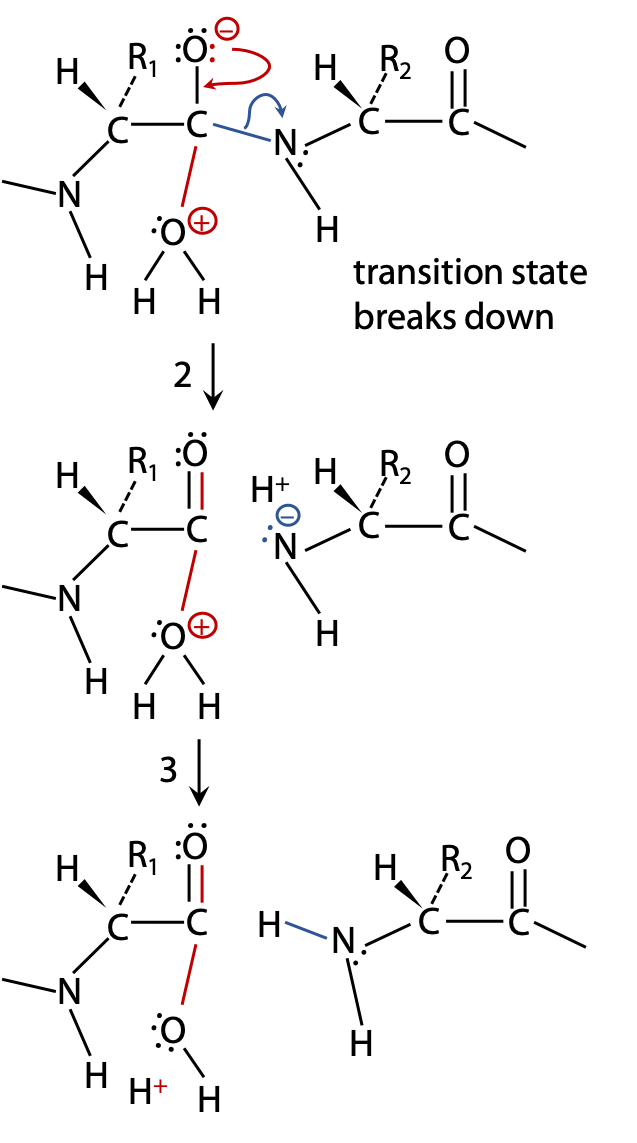
(2) If the transition state breaks up allowing N to withdraw its bonding electrons, the peptide bond is broken. The products stabilize by exchanging protons (Figure 9.3).
(3) to become neutral; additional proton exchanges with the surrounding H2O may then lead to –COOH and +NH2 groups, and the reaction is complete (Figure 9.3).
(2B) If the unstable transition state breaks up by withdrawing electrons back to the original H2O, the reactants are restored without any chemical change having taken place, leading to no outcome (Figure 9.4).

The problem with hydrolysis by neutral H2O:
H2O is not a very good nucleophile because O is too electronegative to share its lone pair electrons. OH– would be much better, but not enough is available at normal pH. H2O is also a good leaving group, so the transition state may go back to starting materials and thus, not lead to a net reaction (Figure 9.4). As a result, the uncatalyzed reaction is excessively slow, about one reaction every 10 years.
At high pH, OH– will be a better nucleophile, while at low pH, H+ stabilizes the transition state and makes the N into a better leaving group by adding H+. However, a biological catalyst must be able to function close to pH 7.
Chymotrypsin makes a multi-pronged attack to give about 40 reactions per second (4 x 109 fold increase in reaction rate)
Chymotrypsin uses a better nucleophile in the form of the catalytic triad (Figure 9.7): Asp 102, His 57, Ser 195 side chains.
The transition state is stabilized by the oxyanion hole – Gly 193 and Ser 195 peptide backbone -NH- groups.
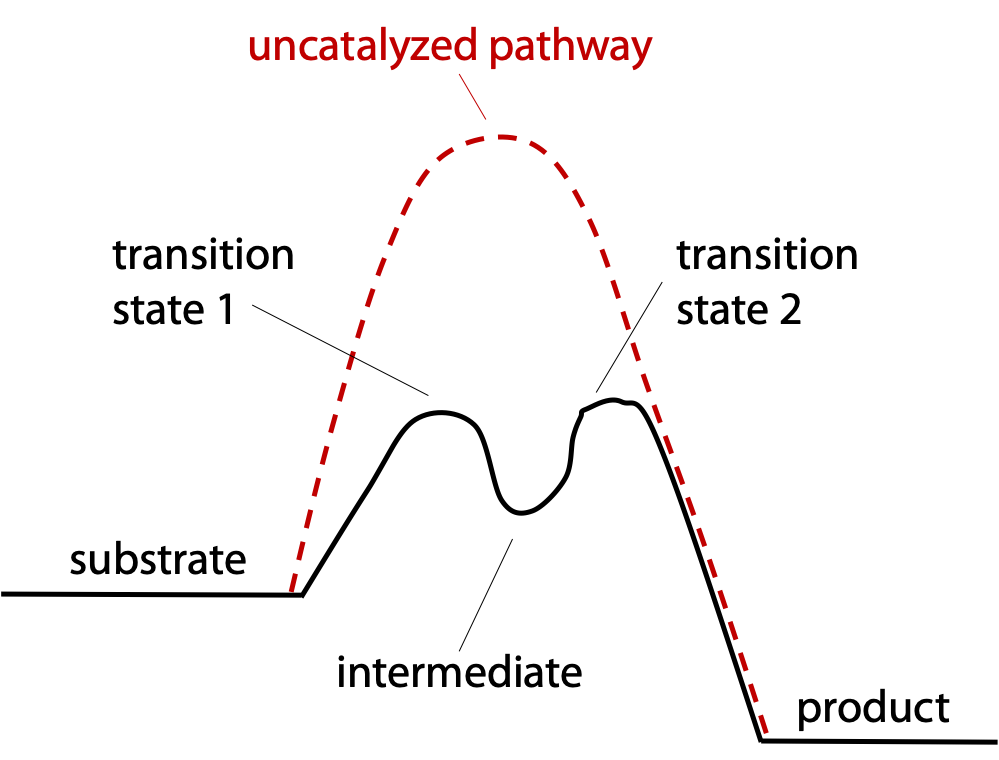
The reaction is taken in two easy steps instead of one difficult step (Figure 9.5).
Step 1: a nucleophile in the enzyme attacks the target peptide bond, splitting off the C-terminal half of the substrate peptide. The N-terminal half remains covalently bonded to the enzyme to form a reaction intermediate (Figure 9.6).
Step 2: The enzyme recruits H2O to split off the N-terminal half of the substrate, restoring the enzyme to its original state.
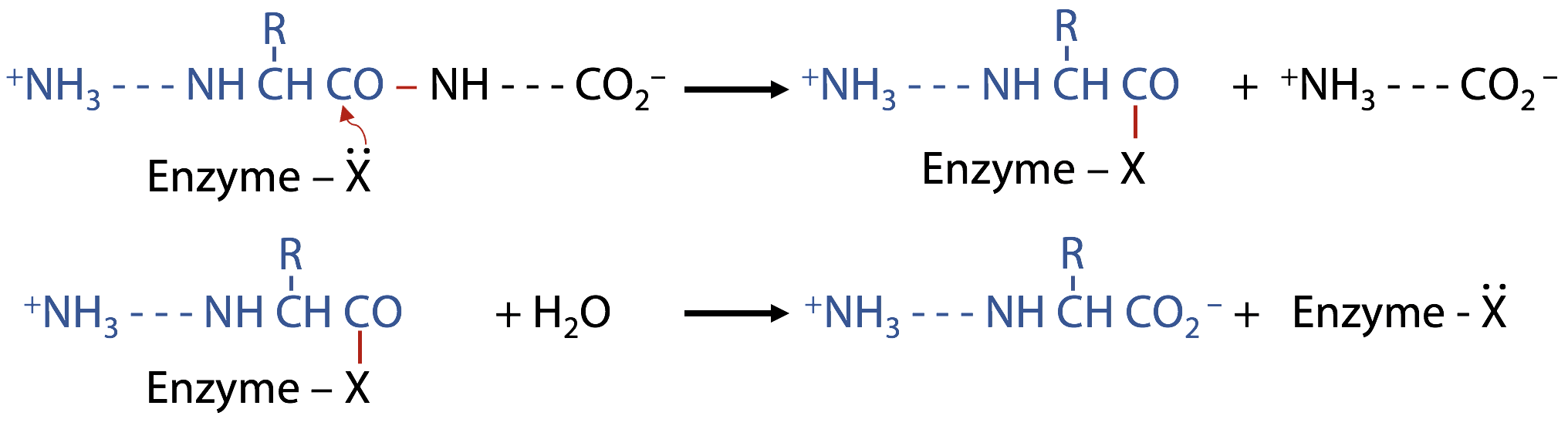
The catalytic triad

The Catalytic triad (Figure 9.7) consists of 3 amino acid side chains, far apart in the polypeptide, but brought physically close together by the folding of chymotrypsin:
1. Aspartate 102 is a negative carboxylate, isolated inside the protein away from external aqueous solution at chymotrypsin surface; it would like to have a +ve charged partner.
2. Histidine 57, a weak base, could be positive but is only weakly protonated since its side chain has pKa = 6.5.
3. Serine 195, side chain CH2-OH, is not a good nucleophile unless it can get rid of the proton, but by itself this does not happen.
The combined action of the catalytic triad is to make Ser 195 into a better nucleophile.
Step 1 of the catalytic reaction (Figure 9.8) (See also Stryer, Fig. 9.7 )
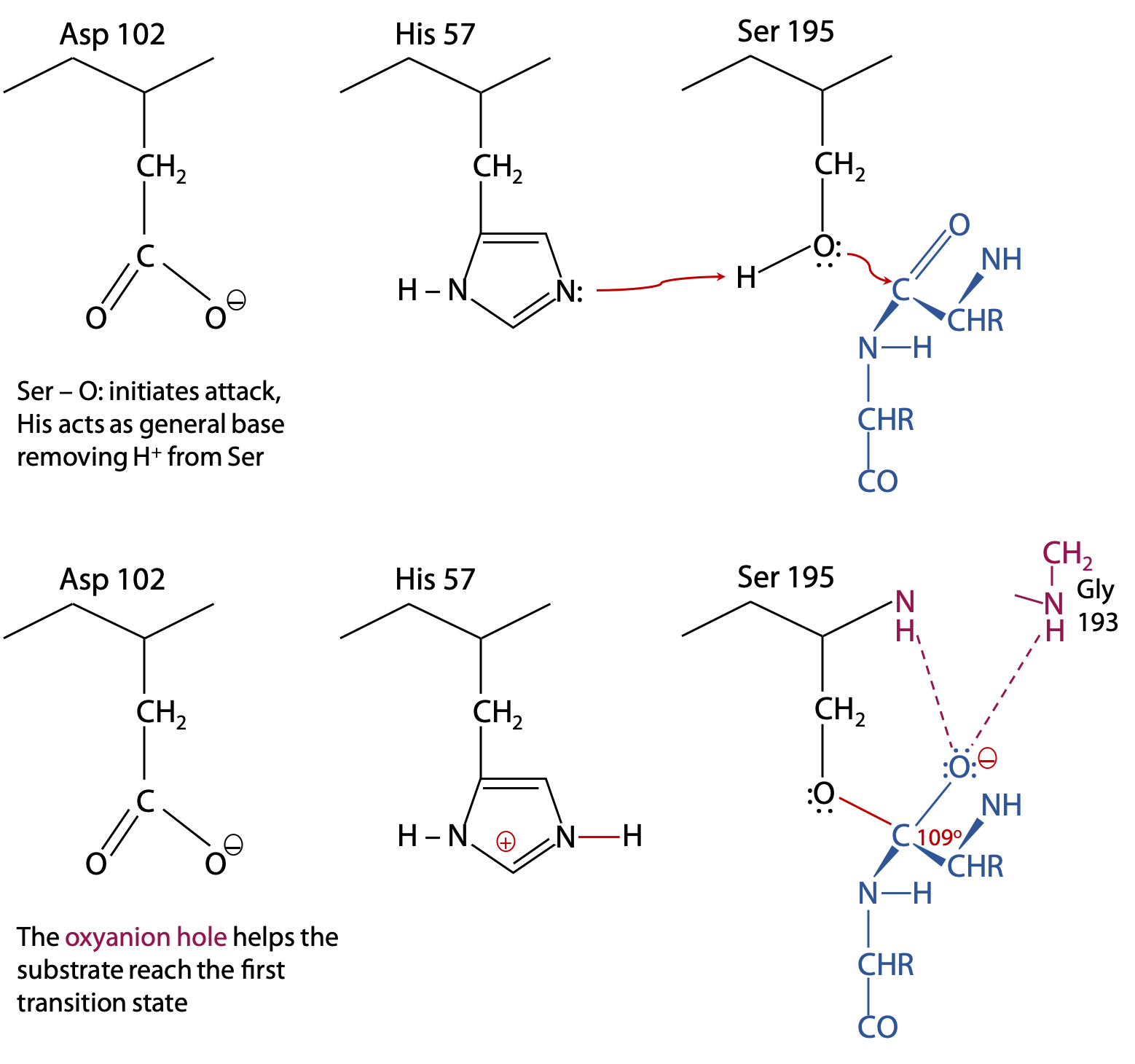
The substrate binds to the enzyme with its target C=O group next to Ser 195. Substrate is shown in blue. The catalytic triad, which is part of chymotrypsin, is in black. Electron exchanges are in red. Cooperative action among the catalytic triad helps form the first transition state.
His 57 acts as a general base by removing a proton from Ser-OH. This helps the Ser O: make its nucleophilic attack on the substrate C=O, while negative Asp 102 promotes formation of HisH+.
The oxyanion hole is also part of chymotrypsin, and consists of the peptide backbone -NH- groups of Gly 193 and Ser 195 (shown in pink). The N-H groups are positioned in such a way that they can donate strong H-bonds to the substrate C=O, but only if the C atom is tetrahedral, as found in the transition state. This strains the bonds of the trigonal planar C=O of the original substrate, helping the reaction to proceed to the transition state.
Breakdown of the first transition state and formation of the intermediate (Figure 9.9):
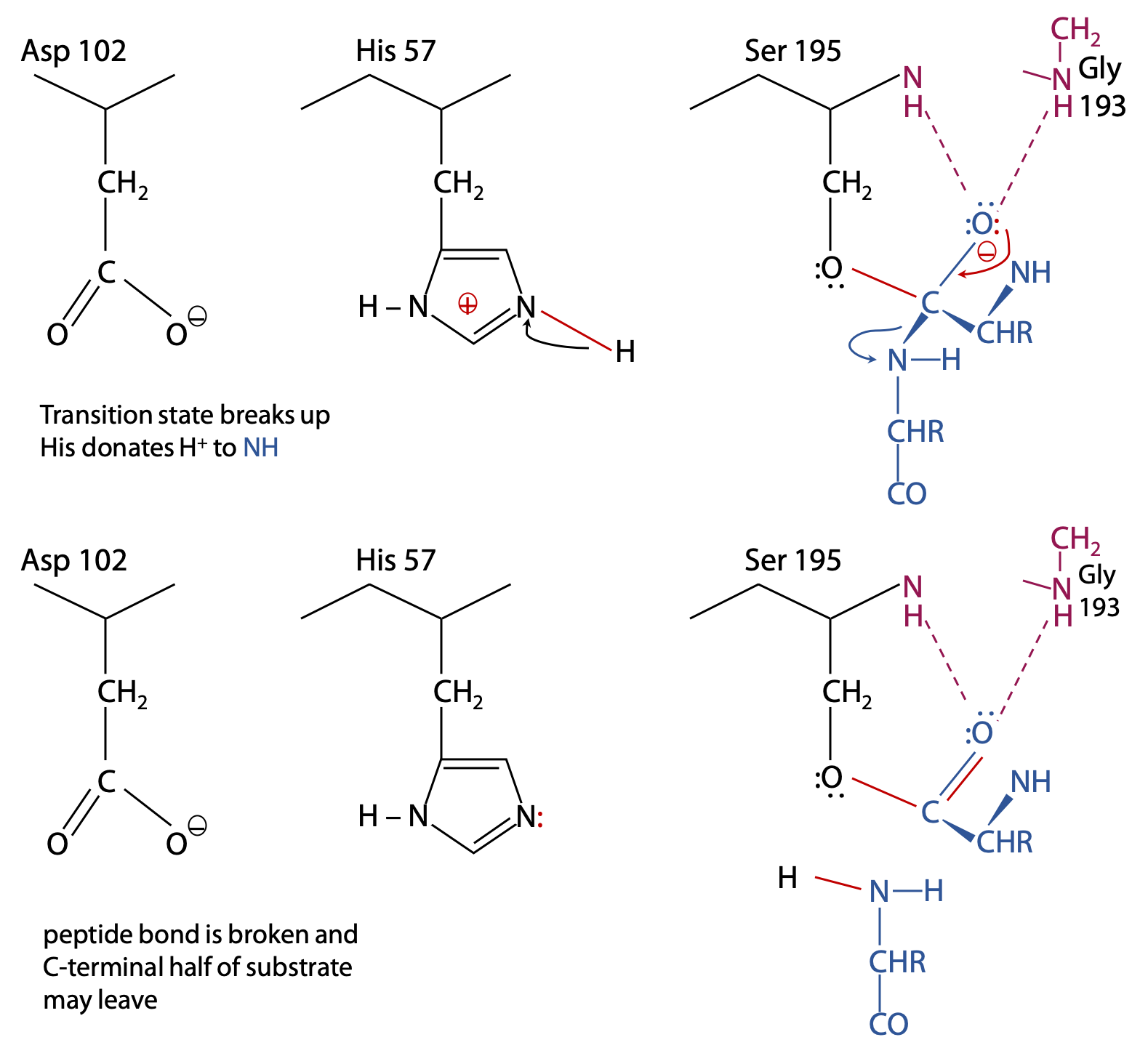
The transition state is broken up by cleavage of the peptide bond following the target amino acid.
Histidine now acts as a general acid, donating its proton to the N atom so that it is a better leaving group.
After gaining a proton from His 57, a neutral NH2– group is formed, so the C-terminal half of the substrate is no longed bonded to the enzyme and is now free to leave.
The remainder of the substrate remains bonded via its carboxylate group in an ester bond to Ser 195. This is called the acyl-enzyme intermediate.
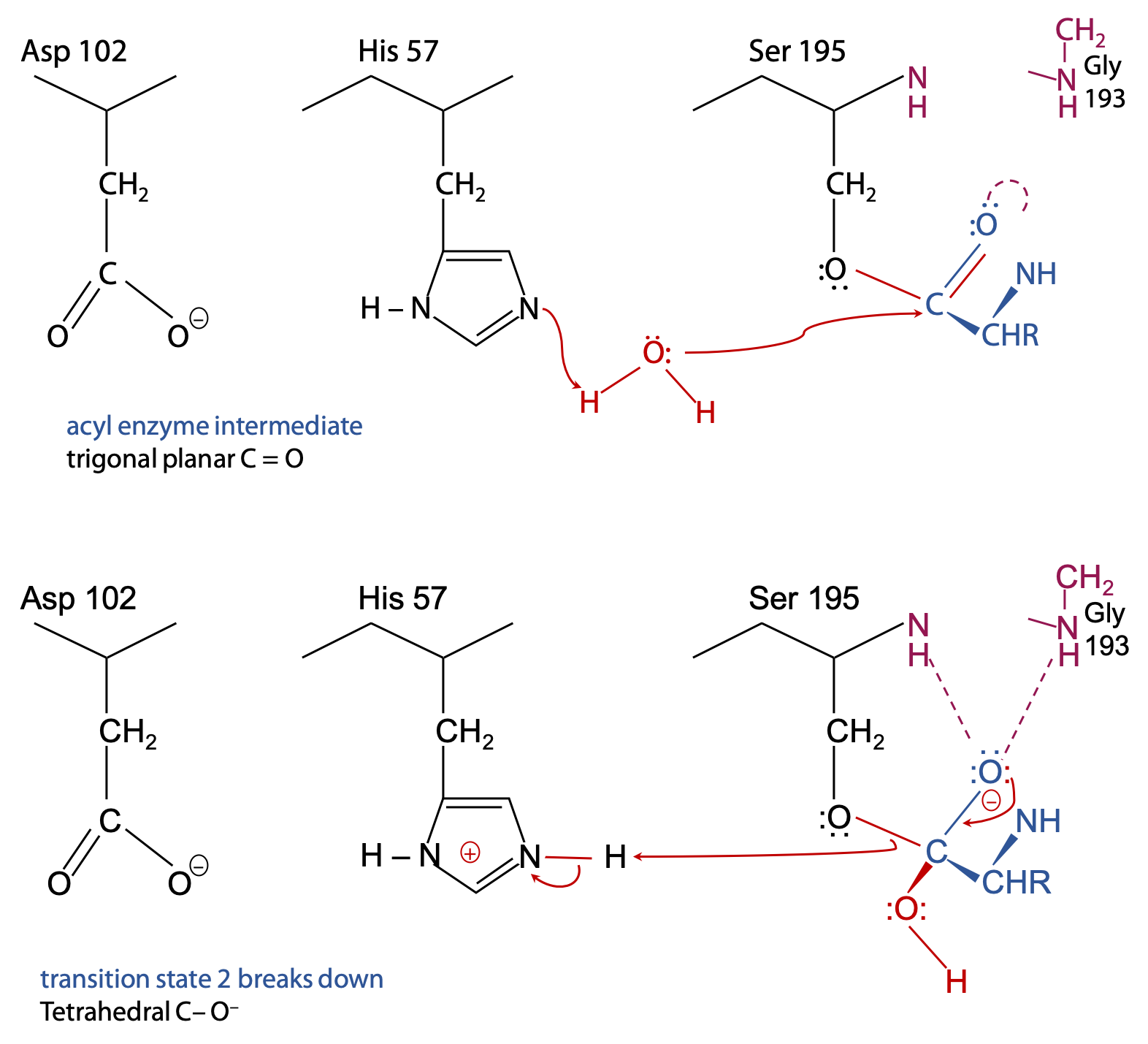
Step 2 (Figure 9.10)
With half of the substrate out of the way, there’s now room for an H2O to get into range of the catalytic triad.
His 57 acts as a general base to remove a proton from H2O, enhancing the nucleophilic power of the O in H2O (much like the earlier step with Ser 195).
H2O can now attack the substrate.
Transition state 2 breaks up because HisH+ 57 acts as a general acid to donate the proton back to Ser 195. It breaks the Ser-O to substrate C-O ester bond.

Final result (Figure 9.11):
The N-terminal half of the original peptide substrate now carries the target amino acid at its newly formed C-terminus. This is free to leave in the form of a carboxylic acid, since the C=O is trigonal planar and pops out of the oxyanion hole.
Some points to note
At the end of the reaction, the catalytic triad is back to the original state. A single enzyme molecule can go through this cycle of reactions with other substrate molecules millions of times over.
The ambivalent behaviour of His 57 is critical to the process. Because the pKa of His is about 6.5, His is able to protonate and deprotonate almost equally well and won’t commit to staying protonated. Deprotonated His 57 acts as a general base, removing the proton from Ser 195 to form transition state 1, and then removing a proton from H2O to form transition state 2.
Protonated HisH+ 57 acts as a general acid donating a proton back to the departing amino group and finally replacing the proton on Ser 195.
This is just an example of an enzyme mechanism. Trypsin and other members of the serine protease family of related enzymes share this mechanism, differing only in the identity of the targeted amino acid. Other enzymes use a variety of chemical processes to bring about the appropriate catalytic process.
Site directed mutagenesis is an important experimental tool for testing theories of enzyme mechanism. A genetically engineered version of the enzyme is produced with a chosen amino acid in the sequence modified. This allows one to test the contribution of each amino acid to the whole process. As one might predict, when Asp 102, His 57 or Ser 195 are changed into other amino acids, this disrupts the catalytic triad and grossly reduces the catalytic efficiency of chymotrypsin.
- Changing Asp to Ala reduces catalytic rate by a factor of 10.
- Changing His to Lys reduces catalytic rate by a factor of 1000.
- Changing Ser to Ala reduces catalytic rate by 106 fold.
Enzymes speed up the rate of a reaction by a definite amount, proportional to quantity of enzyme present.
Enzyme assay is the process or measuring enzyme catalyzed reaction rate.
Enzyme kinetics: mathematical analysis of how the observed reaction rate varies with substrate concentration. Kinetic behaviour can be used to test models of reaction mechanism (rules out wrong models).
Measurement of enzyme rate:

The enzyme is placed above the reaction arrow because it’s a catalyst, not consumed in the reaction (Figure 9.12). The complete reaction cycle restores the enzyme to its initial state.
To measure reaction rate, some property differences between reactant and product must be identified. Rate can be measured as disappearance of reactant or accumulation of product.
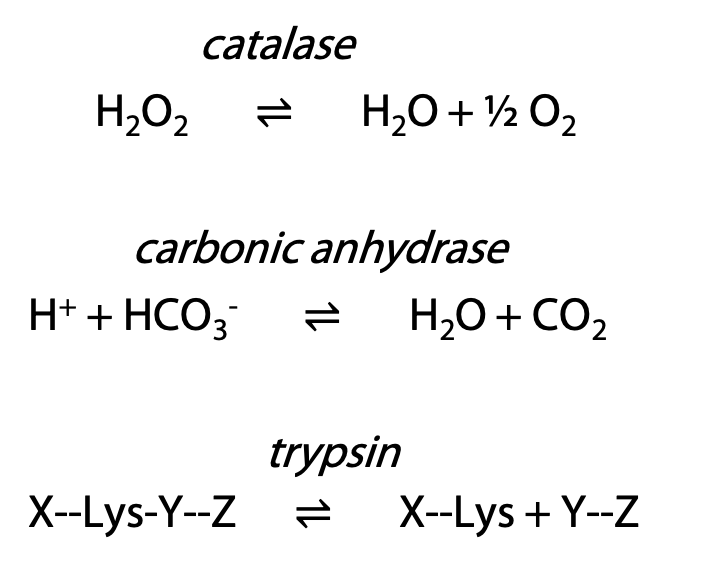
Examples (Figure 9.13):
Measure volume of O2 gas produced.
Measure pH increase as [H+] is consumed.
It’s more difficult when the reactants and products are chemically similar. You could separate the products and analyze each one, but that would be a lot of work in this case.
One approach might be to measure the increase in total reactive terminal amino groups by reaction with the amino acid detection reagent, ninhydrin.
1) Take a sample from the enzyme reaction mixture at intervals
2) Add ninhydrin to each sample: ninhydrin only reacts with N-terminal amino groups, not with peptide-bonded NH groups. The enzyme reaction creates one new N-terminal amino group per peptide bond hydrolyzed.
However, this is not as simple as it looks like.
A more efficient method for trypsin and other hydrolytic enzymes:
Artificial substrate: an artificial substrate is a compound with a structure sufficiently similar to the real substrate that the enzyme is fooled to bind and react with the artificial substrate. However, it is designed so that one of the products has a distinguishing property. For trypsin, a short peptide ends in lysine; however, the lysine is amide-bonded to p-nitroaniline. Trypsin binds and recognizes the peptide sequence ahead of the lysine, so the nitroanilide bond is accepted as if it was a peptide bond, and it is a target for hydrolysis. Trypsin proceeds to hydrolyze the amide bond between lysine carboxylate and the aniline NH2. The free p-nitroaniline has a distinctive colour, which is easily measured (Figure 9.14).
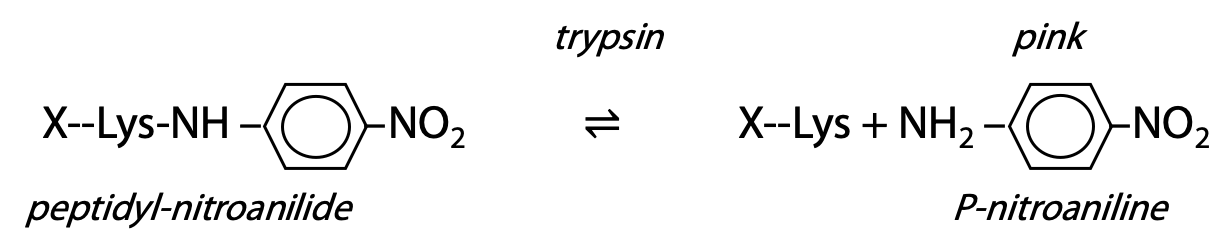
Rate of increase of pink colour is a direct measure of rate of appearance of product.
Artificial substrates are often used for enzymes which hydrolyze simple ester, amide or glycoside bonds.
Some natural substrates show absorbance change on conversion to product:
Lactate dehydrogenase oxidizes the secondary alcohol in lactate to a carbonyl in pyruvate. Lactate dehydrogenase removes 2 H atoms (not H+) from the substrate, hence the enzyme name dehydrogenase. The 2 H atoms are donated to a common biological oxidizing agent, NAD+, nicotinamide adenine dinucleotide, which is reduced to NADH and H+.
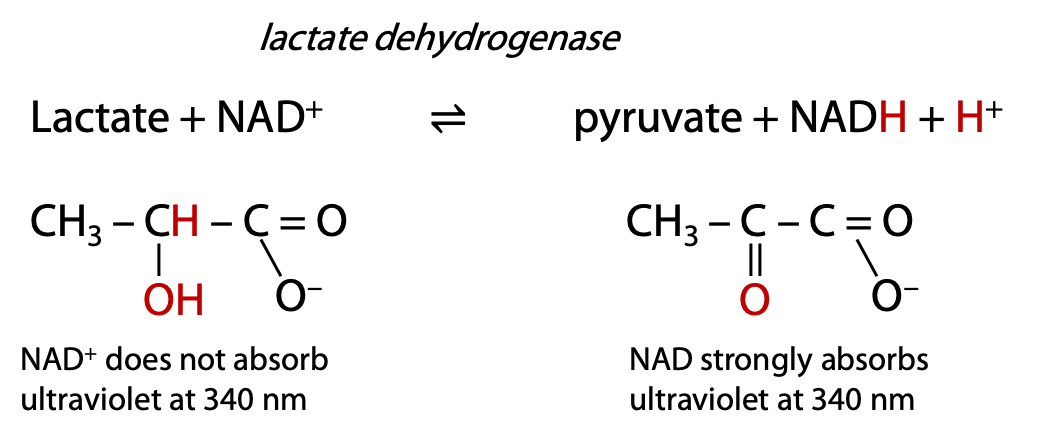
Many metabolic enzymes follow this naming pattern: name of substrate + type of process. The reaction progress can be monitored by measuring the ultraviolet absorbance increase at 340 nm due to the formation of NADH (Figure 9.15).
Coloured or Ultraviolet absorbing molecules possess chromophores.
A chromophore is a part of a molecule that contains conjugated double bonds, a series of at least two double bonds that alternate with single bonds, e.g. N–C=C–C=O. The chromophore absorbs visible (400-700 nm wavelength) or ultraviolet light (200-400 nm). Compounds that absorb visible light appear with a colour opposite to what’s absorbed, so a compound that absorbs blue appears yellow (white minus blue = yellow). Relatively, few biochemical substances absorb visible light, so they are mostly colourless, but several key compounds absorb ultraviolet.
Larger chromophores with many conjugated double bonds tend to absorb at longer wavelengths for example, heme, the red pigment in blood (Stryer, Fig. 24.34).
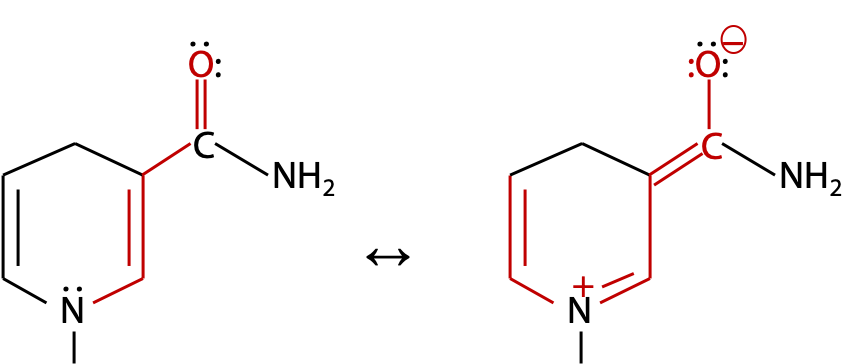
NADH has a relatively small chromophore, so absorbs ultraviolet at 340 nm. The alternative resonance form extends the chromophore through the N atom to the third double bond (Figure 9.16).
Absorbance measurement for ultraviolet or visible light.
LABORATORY-RELATED CONTENT
 The information discussed in this section is related to Lab 2.
The information discussed in this section is related to Lab 2.
Absorbance is measured in a spectrophotometer, which provides a light source with selectable wavelength in the range 200-700 nm (ultraviolet and visible light) (Figure 9.17). A sample is contained in a square chamber or cuvet of exactly 1.00 cm thickness. The light passing through the sample is then measured and recorded by a detector.
If light is absorbed by the sample, the measured intensity I passing through the sample is less than the original intensity of the beam Io.
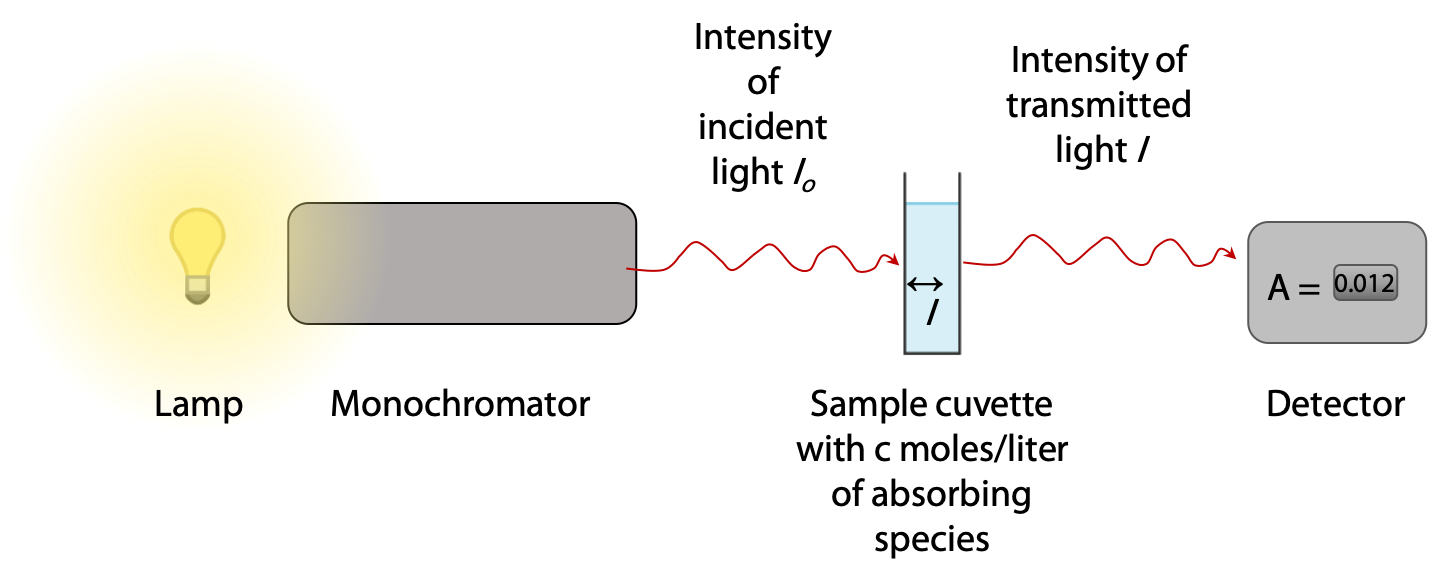
Absorbance A of the sample is defined as A = log10 ( Io/ I ), so if 90% is absorbed, I = 0.1 Io and A = log10 (10) = 1.000. Most spectrophotometers read out directly in absorbance units in the range 0 – 3.000, so the user never has to deal directly with intensity ratios.
The absorbance of a sample is directly proportional to concentration (Beer’s Law) and to sample thickness (Lambert’s Law). When these two relationships are combined, we get the Beer-Lambert equation:
Absorbance A = ε . c . l
where ε = extinction coefficient, a characteristic constant for a given absorbing substance. For NADH at 340 nm, ε = 6200 L mol-1 cm-1.
c = concentration of NADH in mol / L,
l = thickness of the sample in cm (usually 1.00 cm for standard sample cuvettes).
Sample calculation:
A lactate dehydrogenase reaction gave an increase in absorbance of 0.357 units due to the NADH formed.
![\(Increase\ in\ [NADH]=\frac{absorbance\ increase}{\varepsilon.l}=\frac{0.357}{6200}=5.76\times10^{-5} mol/L}](https://ecampusontario.pressbooks.pub/app/uploads/quicklatex/quicklatex.com-c9474c297337ba6553710e21b4656b51_l3.png "Rendered by QuickLaTeX.com")