7 Tertiary Structure & Protein Stability
Synopsis: Proteins consist of polypeptide chains that fold up in a highly specific manner. The starting point for folding is the formation of secondary structure, which is a function of the amino acid sequence. Interruptions of the regular secondary structure by “breaker” amino acids create points of flexibility in the backbone. Folding is then largely determined by clustering of nonpolar side chains to form the core of a globular protein. The majority of proteins fold into a few distinct patterns: α-helix clusters, antiparallel β-barrels, parallel α/β barrels and parallel α/β sandwiches. The stability of proteins is dictated by internal non-covalent bonds, namely hydrophobic effects, van der Waals interactions (dependent of exact matching of molecular shape) and by pairing up opposite charges or hydrogen bonding partners on protein. The Anfinsen experiment proved that all of the information required for the final tertiary structure of a protein was encoded in its primary sequence.
Fibrous proteins and globular proteins
The simplest tertiary structure for a protein to adopt is a single uniform secondary structure:
α-keratin is α-helix (see Stryer Figure 3.29).
Fibroin (β-keratin) is antiparallel β-sheet (see Stryer Figure 3.37).
Collagen forms a unique triple helical structure, the collagen helix (Stryer Section 8.6.1). This is not considered a generic secondary structure, since collagen helix depends on a specific repeating sequence -(Gly-Pro-Pro)n-.
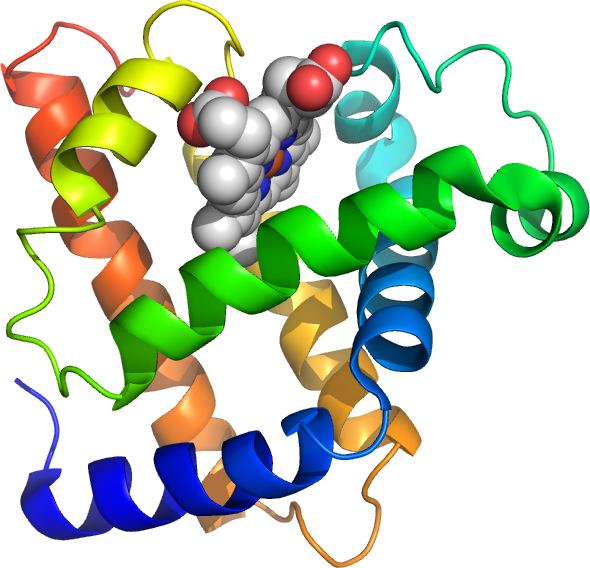
Secondary structures by themselves are rigid and give the protein an overall fibrous shape, whereas most proteins are globular (Figure 7.1).
A globular shape is the result of folding the polypeptide onto itself and depends on the following:
1) Clearly defined interruptions in the secondary structure; clusters of secondary structure breaker amino acids including Gly, Pro, Ser, Asn Asp interrupt rigid segments of secondary structure, create more flexible turn or loop regions where the polypeptide can fold back on itself. Clusters may be pairs of strong breakers (Gly, Pro) or runs with 3 breakers out of four.
A turn is a break of 2-3 residues, often is well defined structure, whereas a loop is a longer section of amino acids with less regular arrangements.
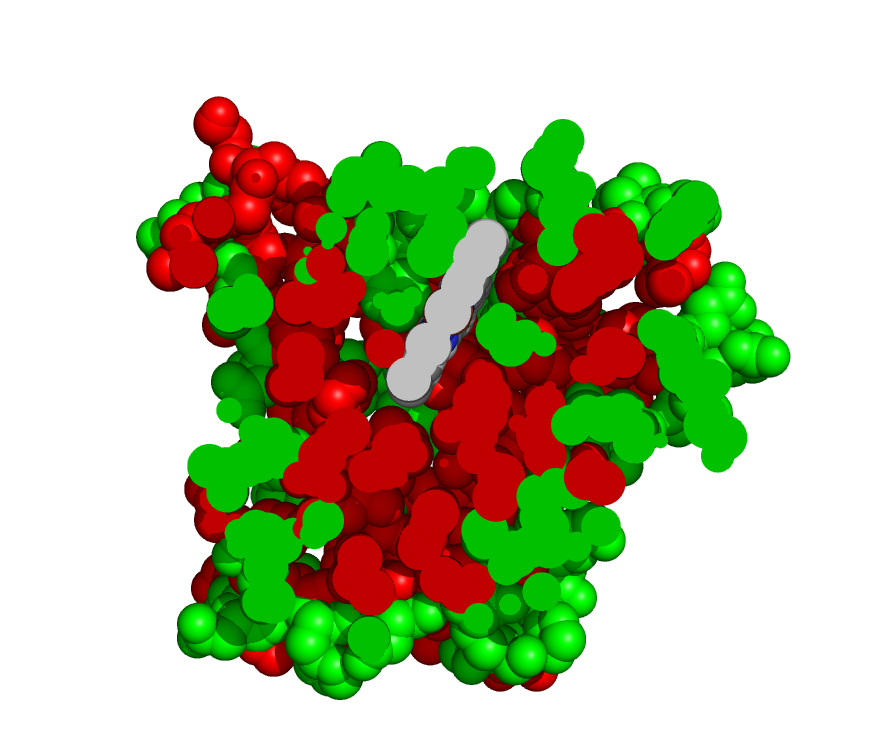
2) The folded protein brings together almost all the non-polar amino acids in the core of the globular shape (Figure 7.2). Thus, the non-polar or hydrophobic amino acids are grouped away from direct contact with H2O. By default the outer shell of a folded protein is largely made up of polar amino acids that interact well with the surrounding H2O. The grouping of non-polar amino acids together by hydrophobic interaction accounts for about 50% of the energy responsible for stabilizing the folded form. The figure shows a cross section through the protein myoglobin, with polar amino acids shown in green and non-polar in red.
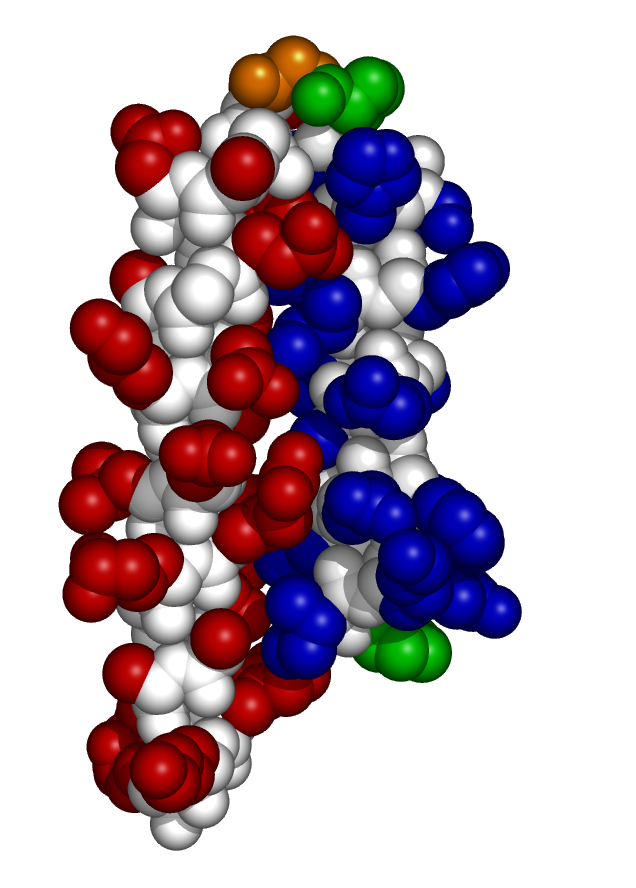
3) The ideal folded state interlocks like a jigsaw puzzle. Figure 7.3 shows two of the helices in myglobin; sidechains on the left helix (red) interlock with sidechains on the right helix (blue).
This arrangement maximizes the number of close atom to atom contacts. Atoms that are in perfect contact bind via a weak attractive force called the van der Waals interaction. This force becomes strongly repulsive if atoms are too close together, and fades to zero if atoms are spaced apart by more than two atom diameters.
Since the van der Waals interaction may be weak (0.1 to 1 kJ compared with 26 kJ for a hydrogen bond or 400 kJ for a covalent bond), the overall effect is significant only if a large number of van der Waals contacts exist, i.e. atom to atom contacts at the ideal distance of separation. This can be true for a correctly folded protein, since a protein molecule contains thousands of atoms in contact.
Improperly folded arrangements don’t achieve this precise fit, so that only a few van der Waals contacts are made at ideal distances. When atoms are too far apart, there is no interaction. When they are too close, there is strong repulsion.
Other stabilizing interactions include:
- Ion pairs, -ve charged side chains paired up with a +ve charged neighbour.
- H-bonds between donor groups such as Arg, Lys, His, Asn, Gln, Ser, Thr, Tyr and acceptor groups such as Asp, Glu, Asn, Gln, His, Ser, Thr, Tyr.
- Disulfide bonds, which form between pairs of Cys side chains that are align side by side the folded protein (more on this in the next lecture).
Most proteins fold in a limited number of patterns
Protein structures usually arise from simple combinations of secondary structures, sometimes known as supersecondary structures, e.g. helix-turn-helix, β-hairpins, βαβ units.
A sequence with mostly groups of α-helix-forming AAs will fold into an α-helix bundle:

In schematic diagrams, cylinders often represent helices. α-helical segments in the polypeptide are interrupted at intervals by breaker amino acids forming the connecting loops. Often, Pro is found near the N-terminal of a helix while Gly often marks the C-terminal limit of each helix. Inward facing amino acids are mostly non polar and interlock jigsaw puzzle style to hold the bundle together (Figure 7.4). Some ion pairs or H-bond pairs may link adjacent helices along the outer shell of the protein.
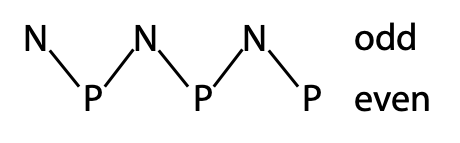
Since the a-helix has 3.6 amino acid per turn or about 7 amino acids for 2 turns, arrangement of polar (P) and non polar (N) in the sequence in a pattern similar to –PNNPPNP- will place all the nonpolar side chains on one face of the helix, leading to the folding shown above.
Bundles of 6-10 helices such as myoglobin (eight helices) appear more complex because the helices tend to splay apart (Figure 7.1).
β-sheet preferring amino acids are not absent, but their distribution is rather scattered, so their “vote” is not effective. α-helical amino acids are more clustered, so their presence determines the secondary structure wherever they are present in a local majority.
A sequence with mostly groups of β-sheet-forming AAs folds into antiparallel β-sheet
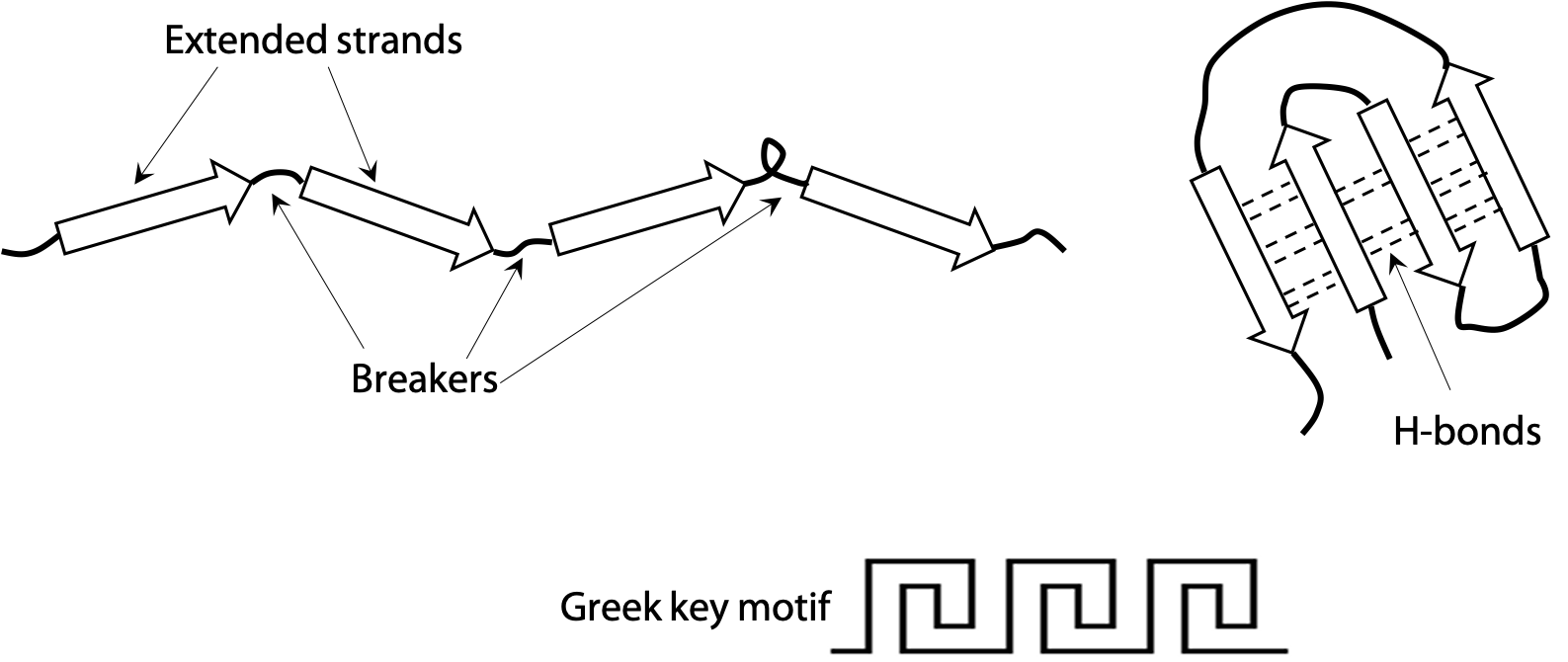
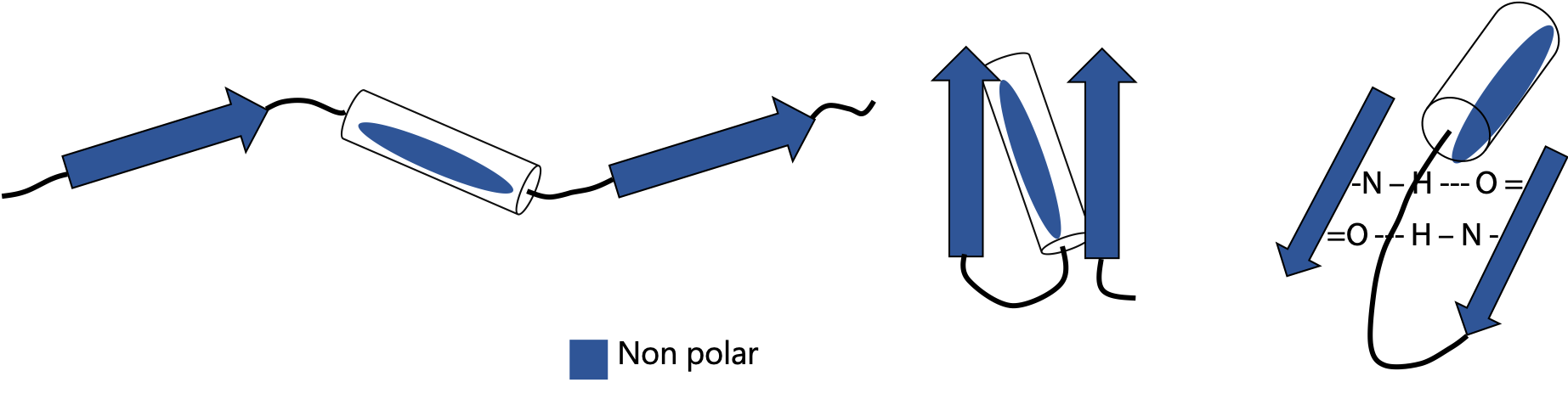
Extended or β-strands are represented by the arrow symbols in Figure 7.5. Two strands connected by a turn naturally tend to form a hairpin structure. A hairpin that flops over gives rise to a four-stranded unit called the Greek key (because it is said to resemble a decorative motif in ancient Greek pottery). Since the strands run in opposite directions in these cases, they line up into an antiparallel β-sheet. Antiparallel is more stable than parallel, due to the good alignment of H-bonds between strands.
Amino acids are frequently arranged to alternate nonpolar and polar side chains. Since a strand has a zig-zag backbone, this places all nonpolar amino acids on one side of the sheet and all polar on the opposite side (Figure 7.6).

The sheet is sufficiently flexible that it can wrap around itself so that the nonpolar side chains face each other on the inside, leaving polar amino acids on the outside (Figure 7.7). An open fold is produced from a sheet of 3-5 strands, but when there are 6 or more strands, the opposite edges can connect up via H-bonds to produce a closed antiparallel β-barrel.
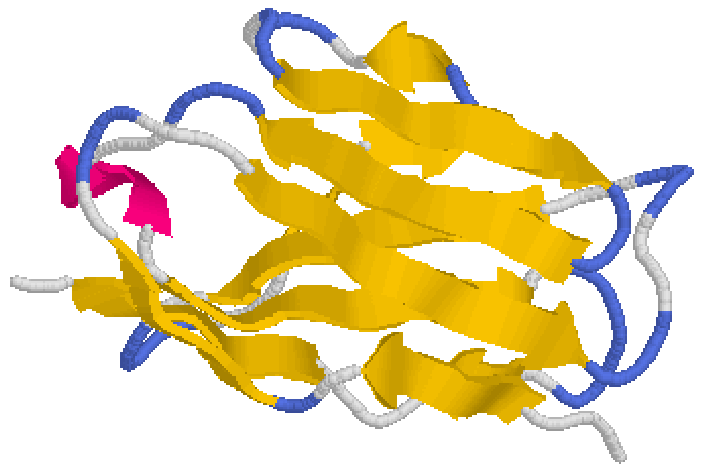
The structure shown is part of immunoglobulin or antibody protein. Yellow arrows are β-strands, blue sections are turns and loops are white. Sometimes a loop may form a short helical segment (red) that is peripheral to the β-sheet core (Figure 7.8). Stryer Section 3.3.
Proteins with alternating Beta-strand and Alpha-helix segments
A parallel β-sheet (extended strands all running in the same direction) can’t form from one continuous polypeptide chain. Instead, when a polypeptide consists of alternating β-strands and α-helix, this allows the polypeptide to run in one direction up the β-strands, and then back down through the connecting α-helix (Figure 7.9).
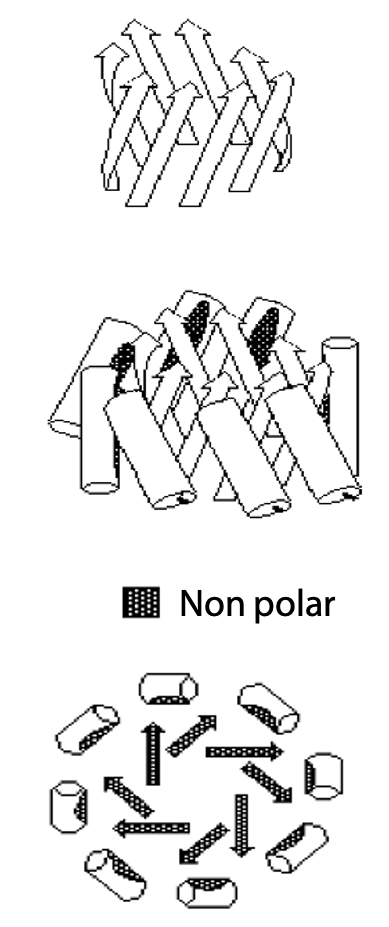
Proteins which alternate β-strands and α-helix can form multistranded parallel β-sheets. The parallel sheet is less stable, due to the angled H-bonds. It is usually totally nonpolar, so the sheet is buried and protected from contact with H2O. If the helices all lie on one side of the sheet, the sheet folds on itself to form a barrel (Figure 7.10). If helices lie on both sides of the sheet, this results in a sandwich structure (Figure 7.12).
A central β-sheet at the core (yellow strands) wraps around to form a closed cylinder or barrel structure, forming a non polar core. This is surrounded by the connecting α-helices (red), for example, the protein triose phosphate isomerase (Stryer, Fig. 16.5).
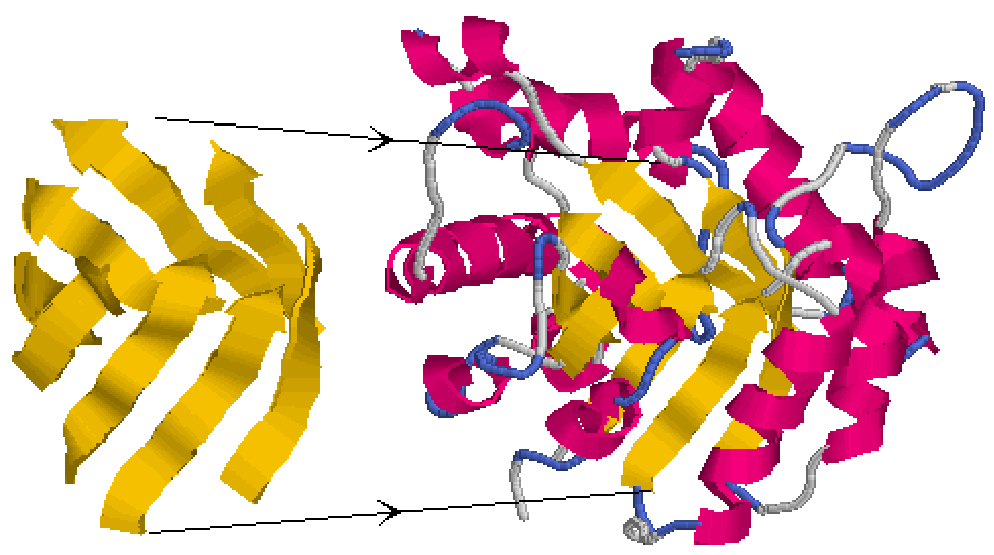
Parallel α/β sandwiches form when the connecting α-helices are arranged on both sides of a central parallel β-sheet. The β-sheet is not flat, instead, is usually twisted, but does not wrap around to form a cylinder like a barrel. The helices cover the central non-polar sheet and protect it from contact with H2O.
Lactate dehydrogenase is an example of an parallel α/β sandwich (Figure 7.12; parallel β-sheet in yellow, surrounding α-helices in red).
a
Domains
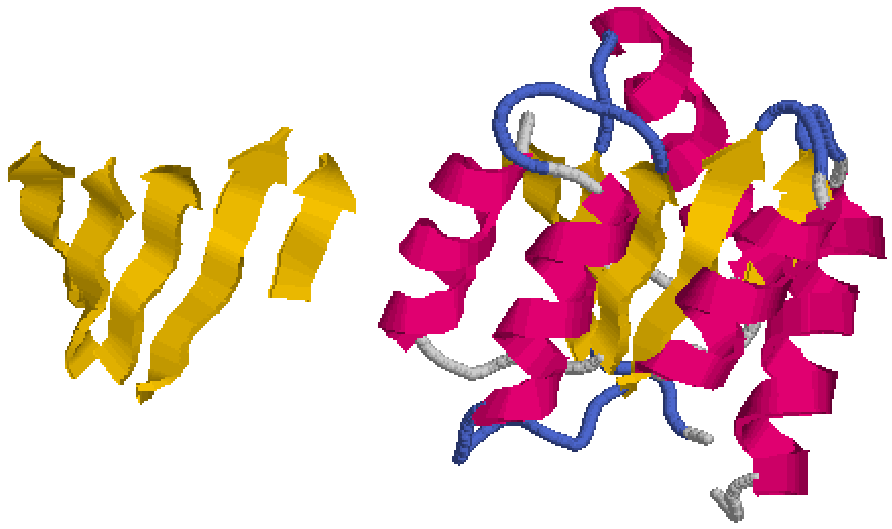
Many proteins may appear more complex, because they are large enough to have several folding units or domains (Figure 7.13).
Each domain of about 10-20 kDa folds up as an independent entity, so a large polypeptide of 50 kDa may be made up of 3 or 4 domains, each with its own folding pattern.
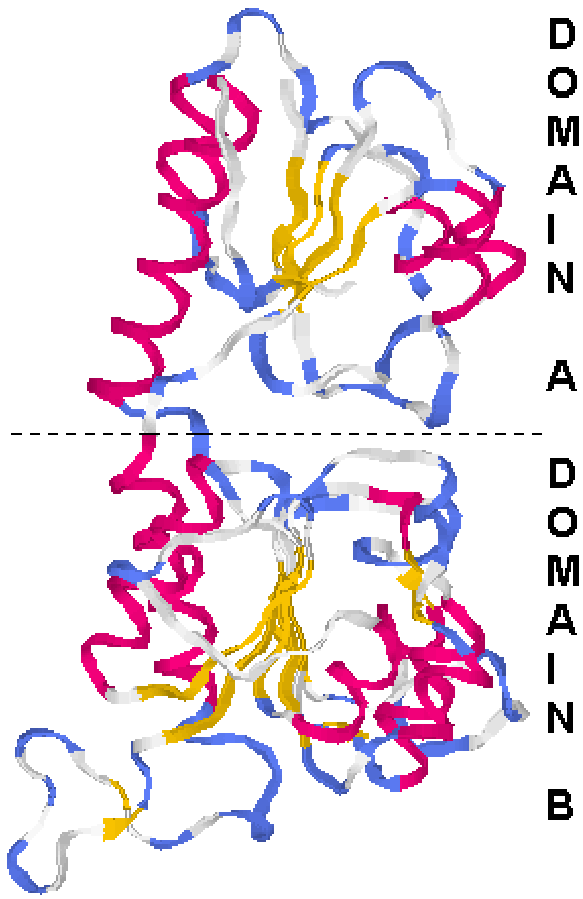
Lactate dehydrogenase folds as upper and lower domains. In this example, both domains happen to be sandwiches, but in many proteins the different domains may have different organizations.
Protein stability
Proteins exist as long polypeptide chains folded into a specific 3-dimensional shape.
The normal folded state or tertiary structure of a protein is called its native state. The exact spatial relationship of the amino acids in the native state give the protein a specific function.
When the polypeptide is unfolded from its normal tertiary structure, the protein is said to be denatured. The spatial relationship of amino acids is disorganized, so a denatured protein loses all of its functions. In some cases, the denatured state may exist as a long extended polypeptide chain (e.g. by the action of sodium dodecyl sulfate in SDS electrophoresis); any shape other than the precise native state is considered denatured and effectively inactive.
Covalent bonding determines the first level of protein structure.
The polypeptide backbone consists of a chain of covalently bonded amino acids linked in a specific sequence – the primary structure.
Non-covalent interactions give rise to secondary and tertiary structures, which dictate the precise pattern of folding and stability of the folded form.
These effects include hydrophobic interaction, van der Waals forces, hydrogen bonding and ionic interactions.
The hydrophobic effect
This describes the tendency of non-polar amino acids to cluster in the core of a protein, out of contact with the surrounding H2O environment. This effect provides the major contribution, about 50% of the total, to the energy that stabilizes the folded state of the protein.
The energy actually arises from strong interactions between polar amino acids and the surrounding H2O. When nonpolar amino acids concentrate in the interior, this maximizes the number of polar amino acids on the exterior, where they interact strongly with the surrounding H2O. Direct contact with nonpolar amino acids has energetically unfavourable effects on the H2O organization (Stryer, Section 1.3.4 & Fig. 1.15).
The magnitude of the hydrophobic effect can be roughly estimated by counting how many CH3-, -CH2– and >CH- groups are moved out from direct contact with the surrounding H2O; each group removed contributes -5 kJ/mol to the overall stability.
The van der Waals effect
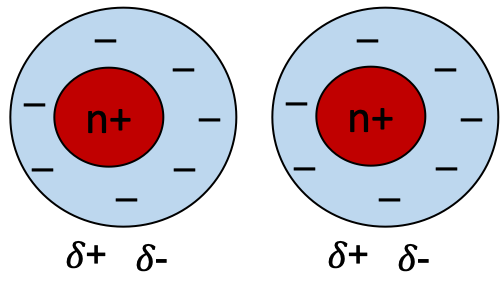
Van der Waals forces are weak electrostatic attractions between atoms which are not covalently bonded, in close physical contact. The interaction arises because of random fluctuations in relative distribution of electrons around a nucleus creates a transient dipole (Figure 7.14). This can induce a near neighbour to become polarised, so they attract each other. The net effect is weak, because the polarisation is temporary. This weak attractive force is called a London dispersion force.
Plot of the energy of van der Waals interaction as a function of distance (Figure 7.15)
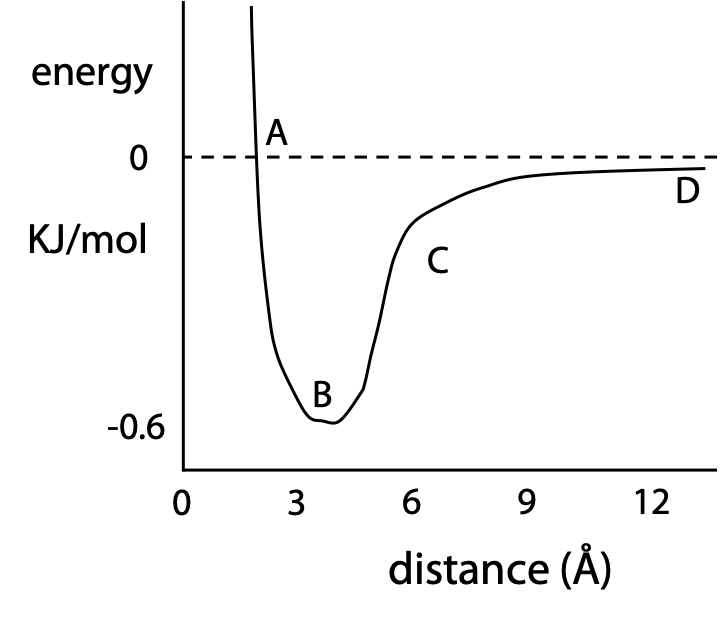
When non-bonded atoms are too close (A), there is a strong repulsion indicated by the steep negative slope and positive free energy. (B) represents the optimum separation distance, about 3.4 Å for a pair of C atoms. (C) A moderate attractive force (intermediate negative energy) exists provided the distance is no more than 2-3 atom diameters, otherwise the attractive force force fades rapidly to zero (D, small negative energy). Although a single van der Waals interaction is very weak, van der Waals forces are significant in protein because thousands of atoms may be in close contact if sidechains in one region are correctly interlocked with a neighbouring region. If the two regions don’t fit each other, only a few atoms come into direct contact, and some atoms may be forced so close that they repel each other (Stryer, Section 1.3.1, Fig. 1.10).
Polar interactions may also help stabilize the folded structure of a protein.

Hydrogen bonds in the polypeptide backbone are important for secondary structures α-helix and β-sheet. Some hydrogen bonds may form between adjacent side chains in the tertiary structure (Figure 7.16).

Ion pairs (sometimes called salt bridges) are electrostatic interactions that result when a positive group, e.g. Lys or Arg, comes in close proximity to a negative group, e.g. Asp or Glu (Figure 7.17).

Ion pairs and H-bonds make less contribution than hydrophobic or van der Waals interactions, because most polar groups face the surrounding aqueous medium. The majority of charged side chains simply pair up with solution ions, e.g. K+ or Cl–, or with dipolar water molecules rather than linking with other side chains (Figure 7.18). The same applies to H-bonding side chains, since H2O is an excellent H-bonding agent. Peptide backbone H-bonds involved in secondary structure generally need to be protected from the surrounding H2O by the side chains of their constituent amino acids.
Ionic and H-bonding groups may act more to destabilize wrongly folded states than to stabilize the correctly folded state.
In the denatured state (unfolded): charged groups stabilize by hydration.
In the correctly folded state: charged groups stabilize by forming opposite pairs or remain hydrated.
In any wrongly folded state : charged groups that become unpaired, dehydrated or wrongly paired (+ve with +ve etc) will decrease the stability of the misfolded form.
Disulfide bonds: covalent contribution to the tertiary structure of some proteins
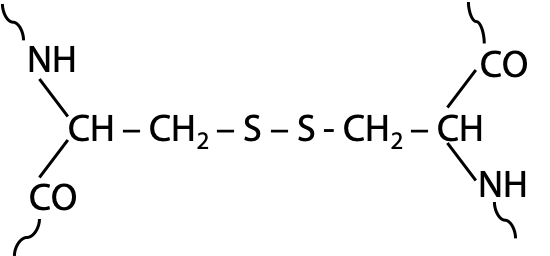
Disulfide bonds may form between pairs of cysteines (-SH side chain), which are physically close in the folded protein (Figure 7.19). Hydrogen is removed from paired –SH groups by reaction with O2. Since disulfides require oxidizing conditions, whereas conditions inside normal cells are often reducing, disulfides are less common than some textbooks may suggest. Disulfide bonds are mostly limited to proteins designed to function outside the cell.
The primary structure of a protein contains all the information needed to specify the normal secondary and tertiary structure
This was demonstrated by Christian Anfinsen in an important experiment in the early 1960’s, to answer the question of how proteins fold (Stryer Section 3.6).
Is some kind of a template needed for the polypeptide to wrap around it, or is the amino acid sequence in the polypeptide sufficient to guide folding with no external assistance?
Starting with the enzyme ribonuclease, a small protein of 124 amino acids, the enzyme is first treated with urea, NH2CONH2. Concentrated urea solutions weaken the hydrophobic interactions, causing the protein to unfold or denature and lose ability to act as an enzyme. In addition, ribonuclease contains four disulfide bonds between cysteine side chains, in specific pairs which are close together in the native state of ribonuclease. The reducing agent 2-mercaptoethanol HS-CH2-CH2-OH is added to reduce -S-S- bonds to individual -SH groups.
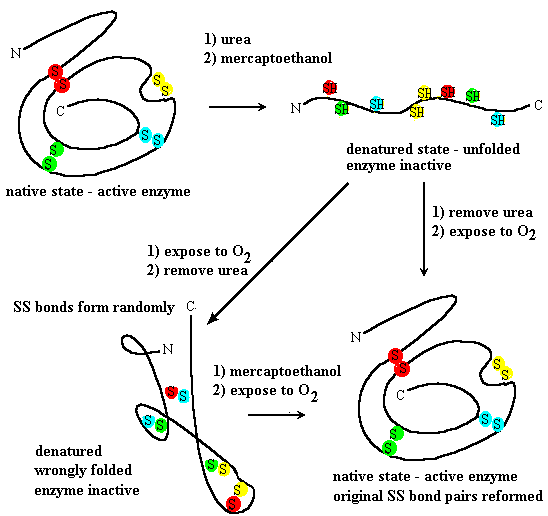
Anfinsen demonstrated that unfolded ribonuclease could spontaneously refold if urea was first removed to allow refolding and then the sample was exposed to air to allow disulfides to re-form (Figure 7.20). The refolded ribonuclease regains enzyme activity, forms the same pairs of disulfides as the original, indicating that the correct tertiary structure formed itself simply from the positions of specific amino acids in the primary structure or sequence.
If the unfolded ribonuclease is exposed to air before removal of urea, disulfides will come together before the correct tertiary structure forms. However, they pair up at random, so the polypeptide can’t arrange itself in the structure needed to behave as an enzyme.