6 Protein Secondary Structure
Synopsis: The polypeptide backbone flexes by rotation about its single bonds. Two arrangements result in regular repetitive structure, α-helix and β-sheet. The particular secondary structure is dependent on the local amino acid sequence. Amino acid can be grouped in three classes: amino acids with bulky side chains prefer to form β-sheet, a second group of amino acids have side chains that disrupt secondary structure and the remaining amino acids tend to form α-helix.
Secondary structure is the occurrence of regular repetitive patterns, such as α-helix, over short sections of the polypeptide chain.
Why is there a pattern?
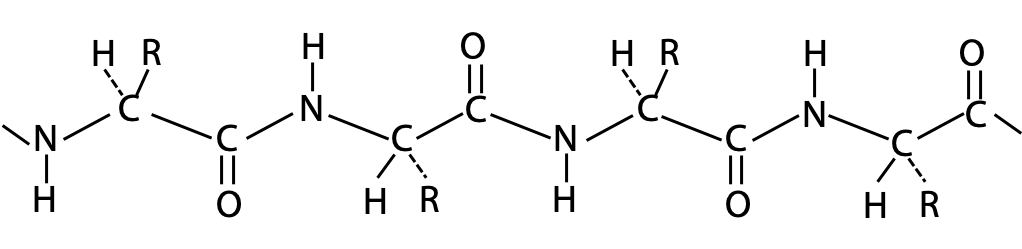
The polypeptide chain forms a backbone structure in proteins. On first inspection, this structure appears to be connected entirely by single C-C or C-N bonds (Figure 6.1). Therefore, it should be as flexible as a simple hydrocarbon chain.

Flexing in a covalent chain structure does not occur by bending bonds, or can be achieved while the normal tetrahedral or trigonal planar bond angles are maintained. Instead, different shapes are obtained by rotation about the axis of single bonds (Figure 6.2).
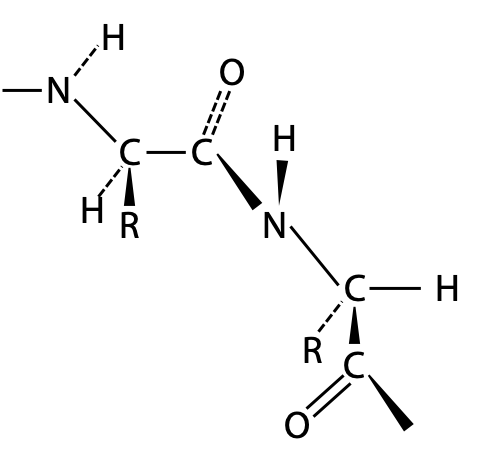
A chain made only of single bonds is highly flexible (Figure 6.3). All polypeptides can adopt a form which is flexible, but it is random and disordered in bond orientation. This is called the denatured state of the protein. Proteins become denatured at elevated temperature or in the presence of disruptive solvents. Because there is no orderly arrangement, denatured protein is non-functional.
Most proteins also have an ordered arrangement called the native state. The organization of the native state places amino acids in 3-dimensional space in the arrangement required for proper function, and this poses the question of why any special patterns should exist at all.
Orderly arrangements of the polypeptide backbone were first studied by examining fibrous proteins called keratins:
α-keratin – protein of hair, skin and wool;
β-keratin or fibroin – spider and silkmoth silk.
The keratins were studied by X-ray diffraction, a technique in which X-rays are reflected off a regular repetitive structure such as a crystal or fibre of protein. The reflected X-rays form a characteristic pattern if the repetitive spacing in the sample is comparable to X-ray wavelengths (Figure 6.4). Since X-rays have the same dimensions as atoms and bonds, repetitive features in molecular structure can be detected. If the X-ray wavelength is known, the size of the repeating pattern in the protein can be calculated.

X-ray diffraction is discussed in Stryer Section 27.1.1. However, detailed theory of X-ray diffraction is beyond the scope of this book.
Visible light can also be diffracted, for example, a spectrum of colours can be seen reflected from the back of a compact disk. The music is recorded on the CD as a pattern of closely spaced dots, and the dots are spaced at a similar distance to light wavelengths. Since different wavelengths are deflected at different angles, white light breaks up into a spectrum.
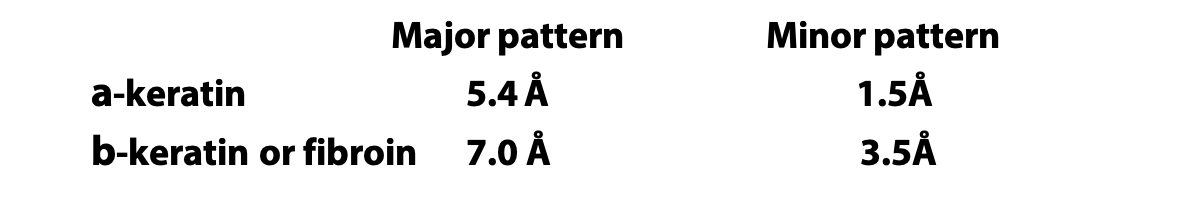
X-ray diffraction gave the following measurements for repeating patterns:
The angstrom unit (Å), 1 x 10-10 meter, is a measure of distance commonly used for atomic structures. For example, the hydrogen atom and the C-H bond are both about 1 Å in size.
The structural basis of these repeated distances were worked out by Linus Pauling. Pauling understood the importance of knowing precise atomic radii, bond lengths, angles and used these to create exact scale models of possible structures.

From bond length measurements, Pauling first worked out that the peptide bond actually behaves more like a double bond, due to the contribution of the second resonance form of the amide (Figure 6.5). This would make the peptide bond rigid and unable to rotate freely, and it can only form distinct cis and trans geometric isomers. In a protein, peptide bonds are almost invariably trans as shown on the left. Bond rotation is allowed only at the α-carbon atoms in the chain.
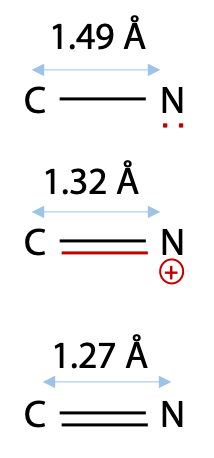
Pauling’s evidence was a comparison of C-N and double C=N bonds with the peptide bond length of 1.32 Å (Figure 6.6).
Since bond length correlates with bond order, this suggested to Pauling that the peptide bond behaves more like a double bond.
At first, Pauling treated peptide bonds as flexible single bonds, but the peptide chain was free to adopt such a variety of structures that no single consistent pattern would emerge. When Pauling included rigid peptide bonds in his models, he found that it limited the number of possible arrangements, so that only certain well-defined patterns would be stable.
Pauling’s models were based on the idea that the >C = N< state of the peptide bond maintains a flat trigonal planar structure which is quite rigid. In the peptide backbone, rigid peptide bonds are linked through the α-carbon atoms, which have tetrahedral shape. Hence, at each α-carbon, the backbone takes a 109° bend. Pauling then found that his models could adopt two basic patterns. Due to the constraints of bond angles and atom size, any other arrangements would force atoms to intrude on each other’s space, i.e. a steric clash:
- a helical state in which the bond rotation at the α-carbon bonds was repeatedly in the same direction.
- an extended state in which the bond rotation at the α-carbon bonds alternated in direction, resulting in a zig-zag structure for the peptide chain.
It is also possible to set the peptide chain in a non-repetitive arrangement called random coil.
The helical form models could be built with varying degrees of twist, but one model fit the atomic dimensions especially well.
Alpha-helix
An α-helix has 3.6 amino acids per turn of the helix. This places the C=O group of amino acid #1 (red O atom) exactly in line with the H-N group (blue N atom) of amino acid #5 (and C=O #2 with H-N #6). The alignment and spacing is ideal for a hydrogen bond C=O:—H-N, and hydrogen bonding makes this structure especially stable. The distance separating each turn of the helix was 5.4 Å, which matches the major repeat distance in α-keratin, hence it has the name α-helix. Since 5.4 Å / 3.6 is 1.5 Å, the α-helix has an amino acid every 1.5 Å, matching the minor periodic repeat of α-keratin (Figure 6.7).
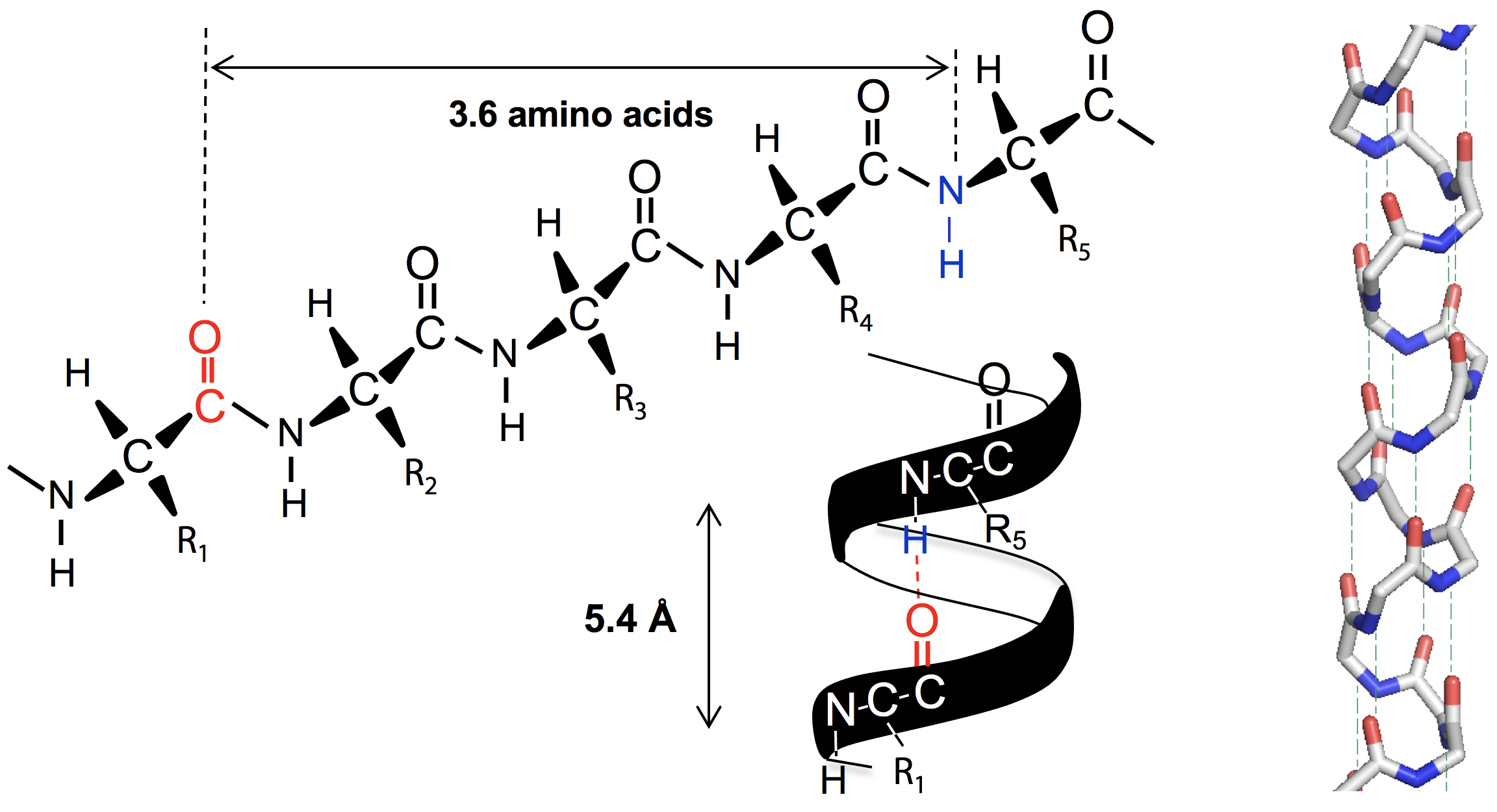
The α-helix was also found to be exclusively right-handed. A left-handed arrangement has similar dimensions, but places the amino acid side chain R too close to the C=O group, making the structure overcrowded. The right handed version of the helix places side chain R next to the N-H group which is much smaller, for a better fit.
In the initial X-ray studies of myoglobin, the same periodic repeats as the α-keratin helix were recognized. The realization that myoglobin was largely α-helix was a major step in solving its tertiary structure. It was the first globular protein to have its full 3-dimensional structure worked out.
Beta-Strands and Sheets
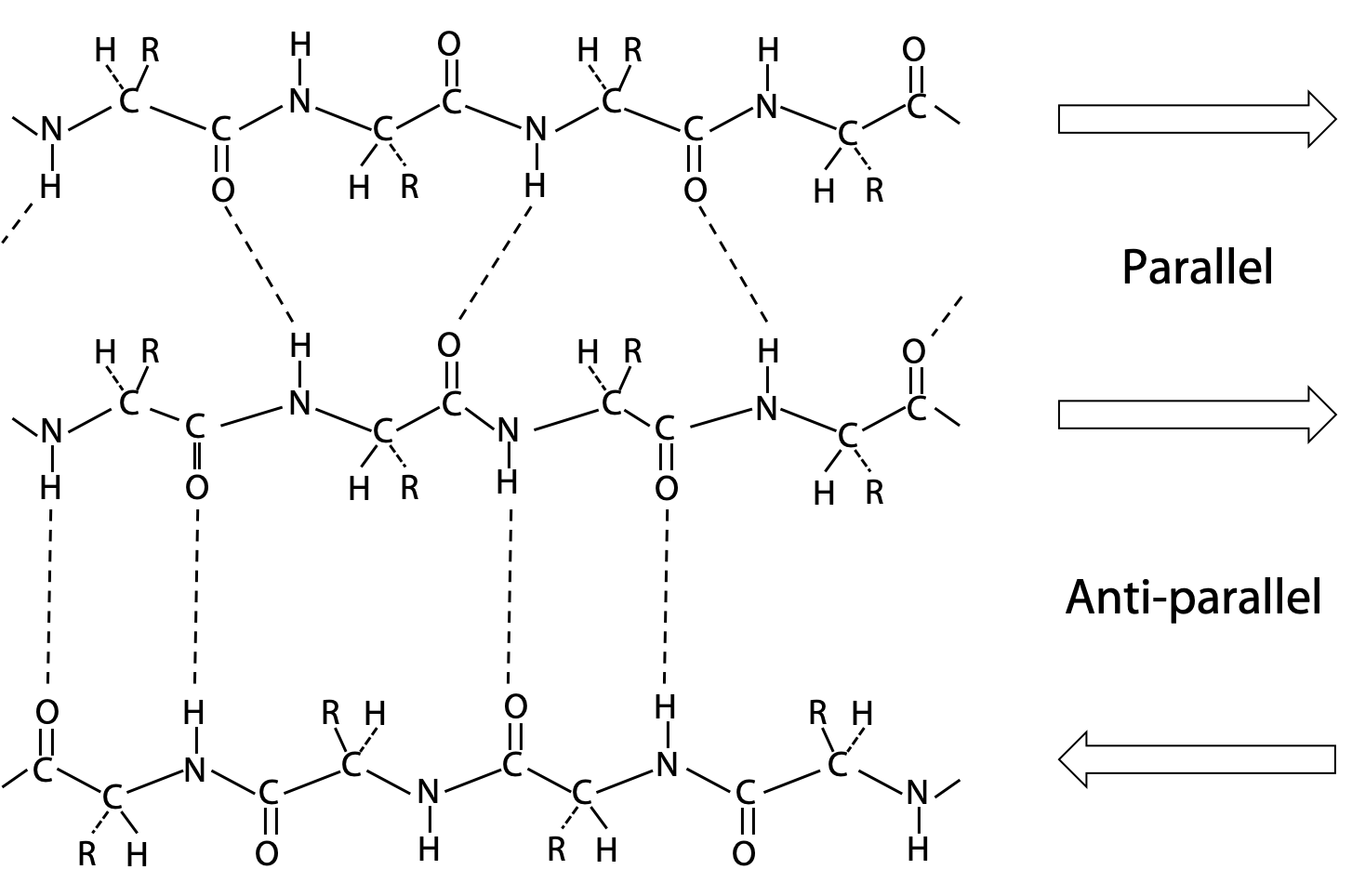
Pauling’s extended state model matched the spacing of fibroin or β-keratin exactly (3.5 Å and 7.0 Å). In the extended state, H-bonding NH and CO groups point out to each side. By lining up extended strands side by side, H-bonds bridge from strand to strand. H-bonds may link extended strands lined up in parallel (same direction) or antiparallel (opposite directions) orientations.
Arrangements with multiple strands form a two dimensional structure is called a β-sheet, because they form the basis of β-keratin structure.
The alignment of H-bonds is much better in the antiparallel arrangement (Figure 6.8).
When the β-sheet is viewed from the edge instead of face-on (above), a striking zig-zag pattern is seen (Figure 6.9). This shows how the β-sheet structure gives bulky amino acid side chains the maximum space and freedom of movement. Also note that odd-numbered amino acids appear on one side of the sheet and even-numbered on the opposite side.

Different amino acids prefer particular secondary structures
β-preferring amino acids: The extended structure leaves the maximum space free for the amino acid side chains. As a result, those amino acids with large bulky side chains prefer to form β-sheet structures (Figure 6.9):
Tyr, Trp (sometimes Phe) are just plain large.
Ile, Val, Thr are bulky and awkward due to branched β-carbon.
Cys has a large S atom on β-carbon.
The β-carbon atom is the first atom on the side chain, so bulky groups are crowded close up to the backbone. The presence of bulky groups on the second carbon of the side chain (γ-carbon), e.g in Leu, is less of a crowding problem.
α-helix preferring amino acids: The main criterion for α-helix preference is that the amino acid side chain should cover and protect the backbone H-bonds in the core of the helix from disruption by the surrounding H2O.
α-helix preference: Ala, Leu, Met, Phe, Glu, Gln, His, Lys, Arg.
Secondary structure breakers: The remaining amino acids have side chains that disrupt secondary structure: Gly, Pro, Asn, Asp, Ser
In the case of Gly, the side chain is a single H atom, which is too small to shield backbone H-bonds from disruption by the surrounding H2O.
Proline is unique because its side chain is directly linked to the backbone N, obstructs the space where the H-bond would otherwise form.
In the case of Asp, Asn and Ser, the side chain is at an ideal length to form H-bonds with adjacent backbone N-H or C=O groups. As a result, these amino acids actually disrupt adjacent H-bonds instead of protecting them.
Clusters of breakers give rise to regions known as loops or turns which mark the boundaries of regular secondary structure and serve to link up secondary structure segments. There are various schemes that give the amino acids numerical weights or rankings for their preferences. Several computer programs can predict the secondary structure from the given sequence (see Stryer, Table 3.3).
Amino acids select a secondary structure by consensus, not individually. If you have a few amino acids that prefer helix randomly scattered in a majority of β-sheet formers, the structure adopted is entirely β-sheet. A section of polypeptide is likely to form α-helix if it contains 60% helix formers in runs of 6 amino acids or more, and no more than 20% breakers. A section of polypeptide is likely to form β-sheet if it contains 60% β-sheet formers in runs of 5+ amino acids, and no more than 20% breakers. 2 or more breakers in a run of 4 amino acids are usually necessary to initiate a turn or loop.