4 Amino Acid Separation, Protein Hierarchy & Chemical Reactivity
Synopsis: Other methods used to separate amino acids and proteins also rely on the properties of the amino acids and include centrifugation, electrophoresis and mass spectrometry.
Protein structure is very much more complex than any simple organic chemical, but by eliminating detail, a pattern or hierarchy of organization emerges:
Methods for separating protein molecules other than chromatography
1. Centrifugation
In centrifugation, a sample is spun in an ultracentrifuge at speeds from 10,000 to 75,000 rpm, producing a force from 10,000 to 500,000 x gravity. At these forces, individual molecules of proteins are large enough to sediment at a rate determined by their size. By measuring sedimentation velocity, it’s possible to derive the molecular mass. It’s theoretically possible, but time consuming to measure sedimentation rates of proteins down to 10 kDa.
2. Electrophoresis
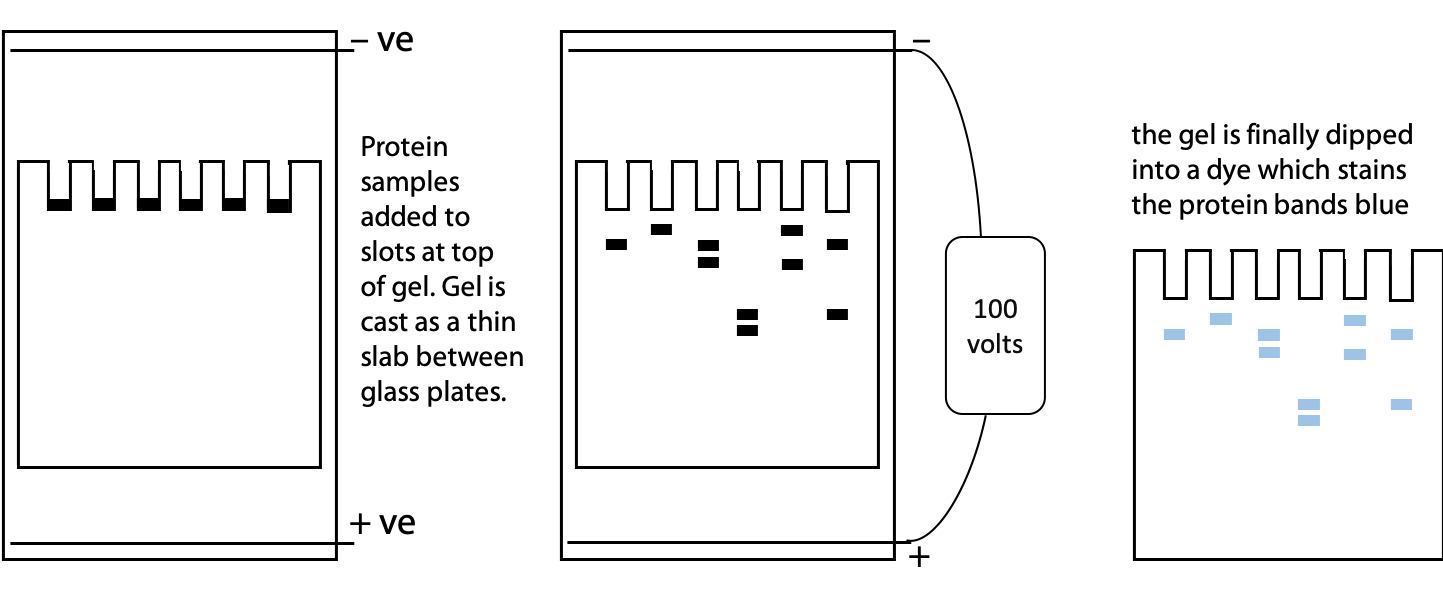
Electrophoresis is separation based on movement of charged particles in an electric field. A mixture of proteins is placed between a pair of electrodes immersed in a conductive buffer solution with a voltage of 100-1000 V applied. Positive molecules move towards the negative electrode and negative molecules move towards the positive electrode. The rate of movement is a function of size, shape and charge. (See Stryer Figure 4.7 for an illustration of electrophoresis apparatus.)
Since free solution is subject to disturbances by convection (local fluid motion caused by temperature differences), the buffer is immobilized in a gel. On the molecular scale, the gel is sufficiently porous to allow protein sized-molecules to pass through. Agarose gels are best for very high mass, especially DNA where molecular masses may be >10 MDa. Polyacrylamide gels are easily formed from simple chemicals in the lab. They are often used for proteins where molecular mass is in the range 10-1000 kDa. A typical polyacrylamide gel is 5-10% polymer, 90-95% buffer.
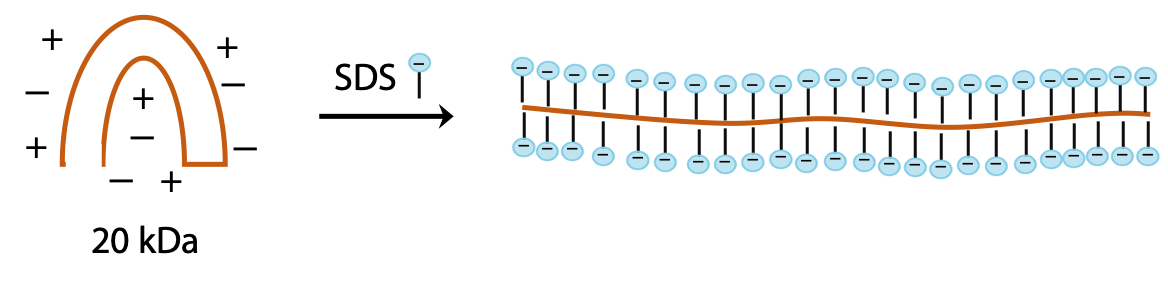
SDS-PolyAcrylamide Gel Electrophoresis (SDS-PAGE)
is a modified form of electrophoresis in which protein is treated with the ionic detergent sodium dodecyl sulfate, SDS. SDS ions coat the protein molecules, which adopt rodlike shapes, so that with SDS bound, all proteins have the same rodlike shape (Figure 4.1). The strong negative charge of the bound SDS ions overrides the somewhat variable charge of the polypeptide itself. The charge of the complex depends on the number of SDS molecules bound, which in turn depends on the size of the polypeptide. As a result, all polypeptides now behave as if they had similar charge per unit length.
The one remaining factor that distinguishes protein in the presence of SDS is the increased frictional resistance for larger proteins. Hence, the separation is based on size, with smaller proteins being most mobile and larger proteins being retarded (opposite to gel filtration).
We can use SDS gel electrophoresis to get information about the size or molecular mass of a polypeptide. In addition to the unknown protein samples, a set of proteins of known size are included in a separate lane of the gel. These proteins are used to create a calibration plot to match distance migrated to molecular mass. This is one of the standard laboratory methods for determining protein size or molecular mass (Figure 4.2). See Stryer Figure 4.7.
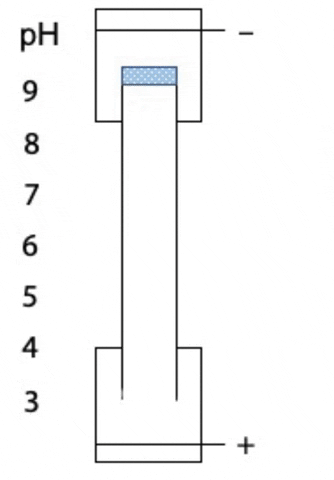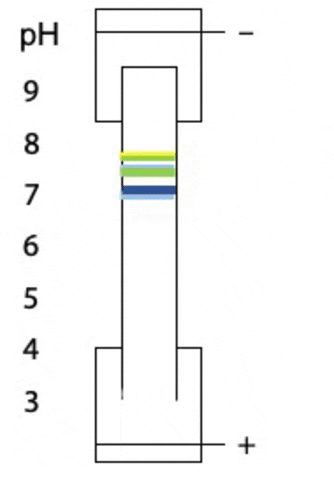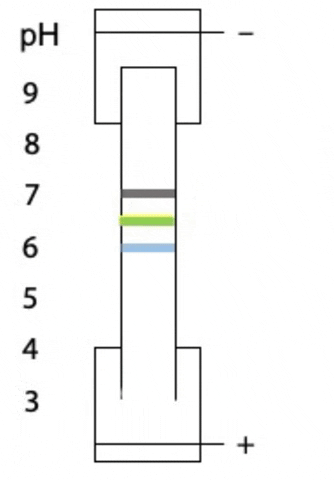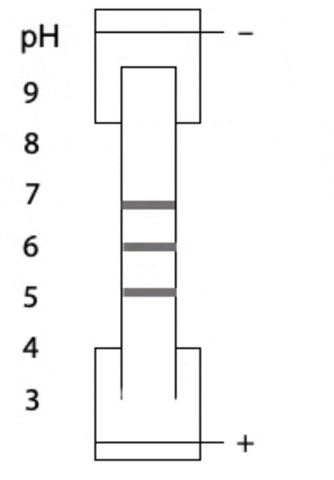
Another variant on electrophoresis is called isoelectric focussing. This is electrophoresis in a pH gradient and separates proteins on the basis of charge.
Every protein has an isoelectric point, the specific pH at which the sum of negative charges is exactly equal to the sum of positive charges and its net charge is zero. If a protein starts at the high pH end of the gradient, it will have negative charge. If the positive electrode is placed at the low pH end of the gradient, the protein migrates towards the positive electrode, passes through buffer of gradually decreasing pH. As the pH decreases, different side chains in the protein become protonated, causing the net negative charge to decrease. At some point, the protein reaches the pH equal to its isoelectric point, where it has no charge and stops migrating, since there is no attraction to either electrode. Separation occurs because each protein in a mixture has a different isoelectric point (Figure 4.3).
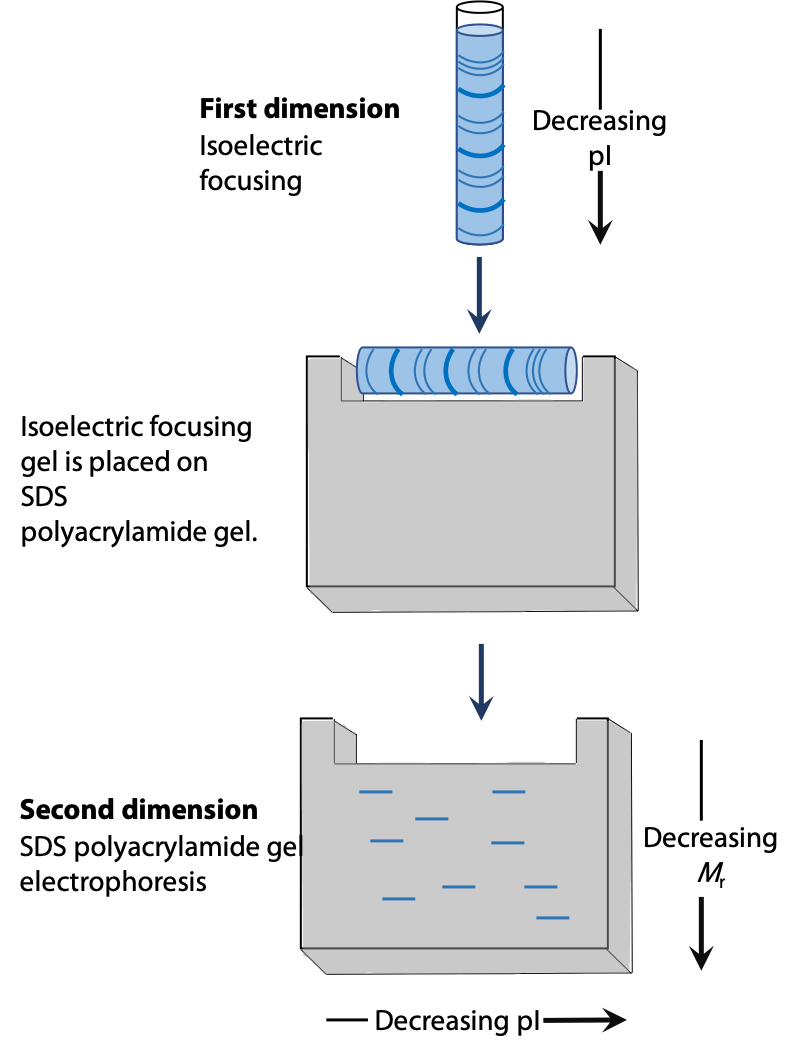
Two dimensional electrophoresis involves separation of a protein first by isoelectric focussing in a thin capillary tube. The spaghetti-like gel contains the partly separated proteins. Then it is laid on the top edge of a conventional SDS-PAGE gel. After that, a second separation by electrophoresis is carried out at 90˚ to the original isoelectric focussing (Figure 4.4). See Stryer Figure 4.12.
Mass spectrometry is a technique often used in conjunction with electrophoresis to identify proteins (Stryer Figure 4.16).
A pure protein sample is obtained as a band cut out from gel electrophoresis. The sample is either introduced into a high vacuum chamber as a superfine spray, or vaporized by laser bombardment on a positively charged electrode. This yields charged protein ionic particles, which are accelerated by attraction towards a negative electrode. A small hole in the negative electrode allows some of the ions to pass through, forming a beam of positively charged protein ions. The velocity of the ions depends inversely on m/z, the ratio of mass to charge. Since the unit charge on an electron or proton is known, the exact mass of the protein can be calculated by measuring the time of flight – the time it takes the beam to travel down a tube of known length from negative electrode to detector. The protein mass can then be compared with a catalog of proteins of known mass.
The laser method is known as MALDI-TOF mass spectrometry, for Matrix-Adsorption Laser Desorption Ionization-Time of Flight mass spectrometry.
Levels of Protein Structure Hierarchy
Primary structure, the specific sequence of amino acids in the polypeptide chain
Secondary structure, the occurrence of regular repetitive patterns over short regions of the polypeptide
Tertiary structure, the overall folding of the complete polypeptide chain
Quaternary structure, the assembly of several protein molecules to form a larger complex with distinct properties
These four levels of protein structure depend in various ways on the amino acid sequence, so the chemistry for determining amino acid sequence forms our starting point for an exploration of protein structure.
___________________________________________________________________________

A protein consists of a long linear chain of amino acids.
Myoglobin, an oxygen binding protein found in muscle tissue, has 153 amino acids in its polypeptide chain (Figure 4.5).
This is a relatively small protein. Some proteins contain hundreds or even thousands of amino acids.
Describing the protein as a set of amino acids is one way to simplify the structure; however, it is not a complete or accurate view of the three-dimensional structure.
Protein structure is clearly very different from that of simple organic compounds.
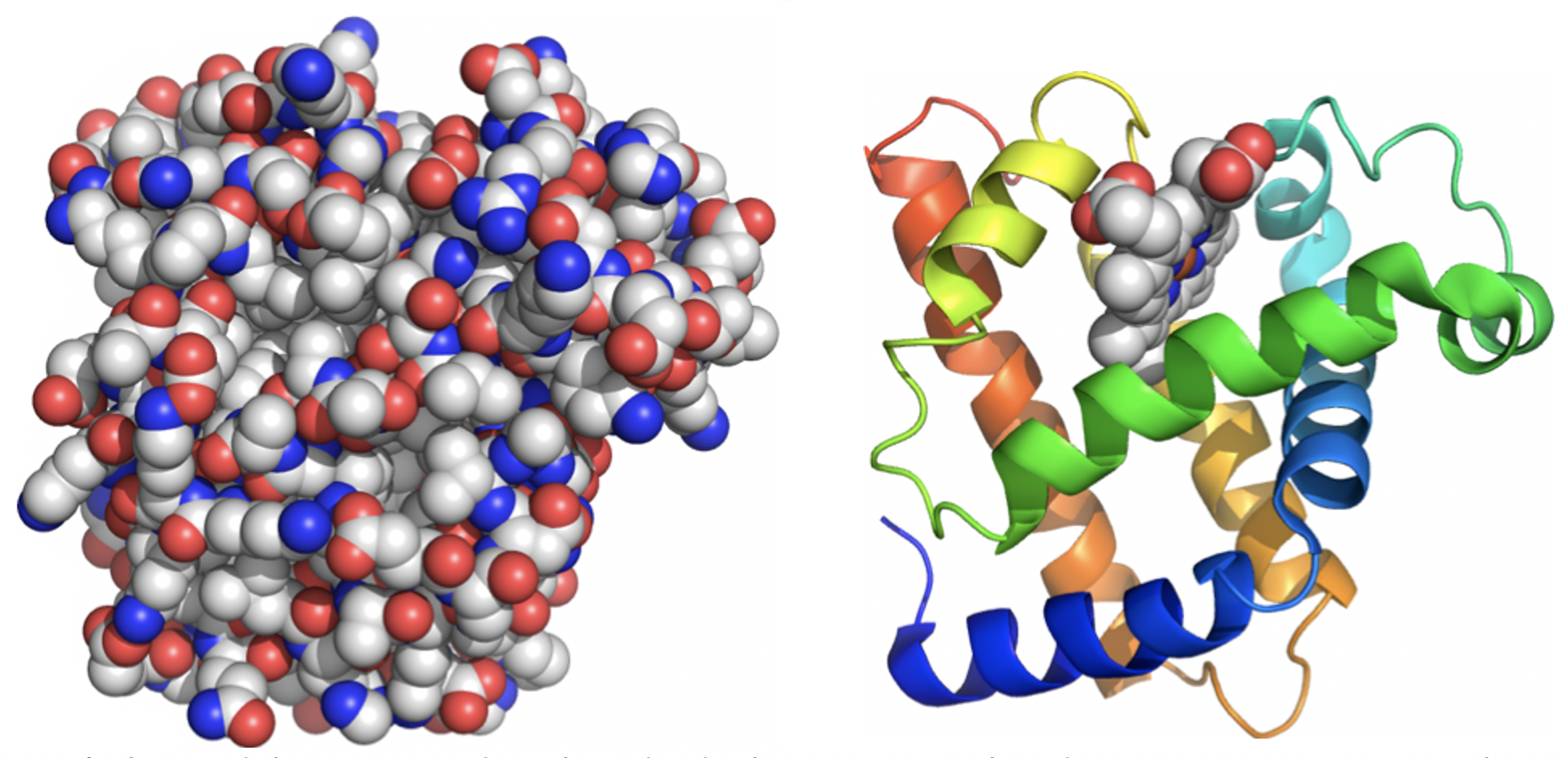
We can’t be unduly concerned with individual C-C or C-H bonds; attempting to view the myoglobin structure at the level of detail shown on the left makes it impossible to grasp the overall organization (Figure 4.6).
Instead, the structure of protein is viewed through a series of simplifications. Computer software can be used to suppress detail and make visual interpretation easier. The structure on the right is the same myoglobin molecule seen from exactly the same viewpoint as on the left. It traces the path of the polypeptide backbone as a ribbon and eliminates the side chains. Colour can be used to distinguish the sequence – the N-terminus is shown in blue and progresses through the spectrum until we reach red at the C-terminal end.
Regular structural organization now becomes clearer. Protein structure can be subdivided into a hierarchy of three or four levels (Figure 4.7):
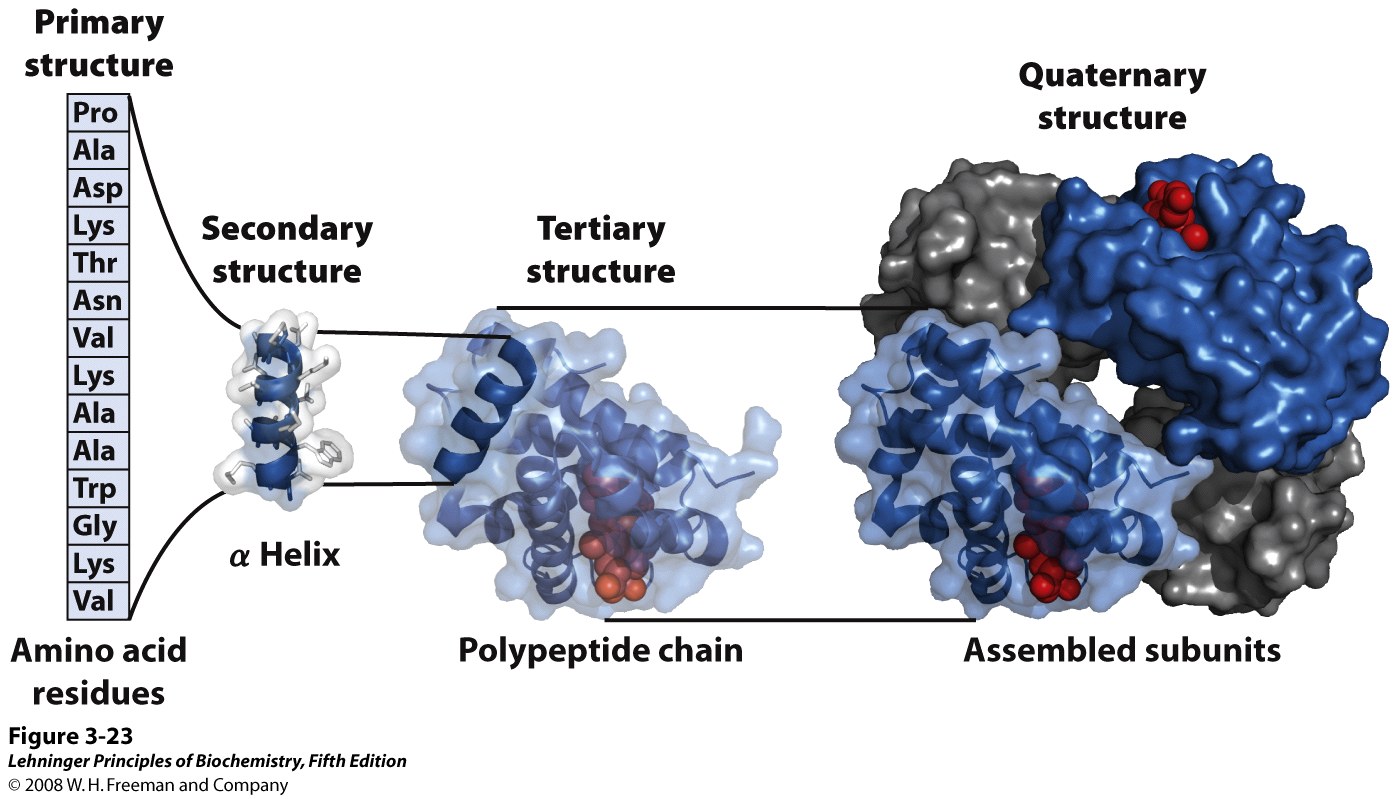
Primary structure is the linear sequence of amino acids in the polypeptide chain. By convention the N-terminal amino acid is considered the start, amino acids are numbered counting from the N-terminal end.
The way that proteins function is dependent on spatial organization and close placement in 3D space of particular amino acids which might be far apart on the linear polypeptide. This means that each protein consists of a polypeptide that folds up in a highly specific manner and the pattern of folding is as important to the structure as the covalent bonds.
Secondary structure: the polypeptide backbone is represented as a single ribbon, N terminus at lower left. The ribbon forms regular helical or spiral patterns in some parts and irregular loops elsewhere. Regular repetitive folding patterns over short sections of the peptide chain (5-20 amino acids long), such as the helix sections appearing in myoglobin, are called secondary structure. (More details on secondary structure will follow in upcoming lectures.)
Tertiary structure is the overall folding of the whole polypeptide. For myoglobin, 8 helical secondary structure segments fold together to enclose a central cavity. (More details on tertiary structure will follow in upcoming lectures.)
Quaternary structure is the assembly of several individual polypeptide chains into a larger structure that has special properties. Hemoglobin, the O2 binding protein of blood, consists of four independent molecules of globin, each similar in size and structure to myoglobin. The globin units associate by non-covalent forces, but behave in a cooperative manner to make the O2 carrying function of hemoglobin more effective. (More details in BIOC*3560.) Only a few proteins have quaternary structure.
Investigation of structure
All higher order structure (secondary, tertiary, etc) arises from the primary structure, namely the amino acid sequence within the polypeptide. In order to find out how a polypeptide chain is made up, we need to find out what amino acids are contained in it and in what order or sequence they occur. To do this, it is necessary to break the peptide bonds (hydrolysis), so that the amino acids can be identified.
Practical aspects of peptide hydrolysis (attack by water)
H2O itself hydrolyses peptides bonds extremely slowly, because neutral O: is a poor nucleophile. Although it has two lone pairs, electronegative O: is less willing to share them than N: or S: .
Hydrolysis of peptides and proteins is usually done with a catalyst:
Acid hydrolysis is done in 6 M HCl at 110˚C. It takes 24-72 hours to get complete breakdown of a peptide chain into single amino acids. Trp is destroyed during this process.
Base hydrolysis is done in 4 M NaOH at 110˚C. It takes 16 hours for complete hydrolysis, but some amino acids are destroyed in strong base.
Hydrolysis may also be carried out by digestive enzymes called proteases (more in upcoming lectures). Enzymes are proteins that have a catalytic function, in this case to hydrolyse peptide bonds.
After hydrolysis, the amino acids present in a protein sample can be detected by chromatography e.g. ion exchange or reversed phase.
Underlying basis of chemical reactions
The chemical basis of the peptide bond-breaking reaction such as hydrolysis is a chemical process called nucleophilic displacement. Since many biochemical reactions that take place in living cells are initiated by nucleophilic attack, we need to understand what a nucleophile is, and why it can lead to peptide bond breakage.
The chemical reactivity of a molecule is a consequence of imbalances in the distribution of valence electrons of atoms. Parts of molecules that are primarily C-C and C-H bonded are well balanced, non-polar and chemically inert. However, where atoms seem to have valence electrons to spare or are electron deficient, or draw electrons towards them, these create imbalances where a reaction may occur as the atoms seek a better arrangement.
A nucleophile is simply an atom with a lone pair of electrons that is available to share with another nucleus. A nucleophile is “nucleus loving”, seeks out other atoms (nuclei) that are electron-deficient. This may be a fully or partially positively charged atom. By sharing the electron pair with another nucleus, a new bond is formed.
Atoms with lone pairs:
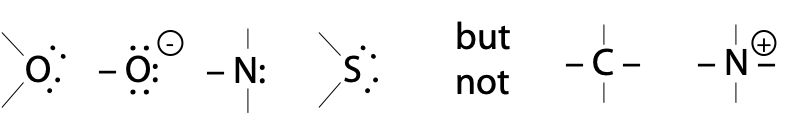
A nucleophilic displacement is a reaction in which an incoming nucleophile X: attacks a target atom C to displace another attached group. The group Y that detaches is called the leaving group:
X: C –– Y → X –– C :Y
An atom with a lone pair can use it in different ways:
1) The atom acts as a hydrogen bond acceptor if it simply attracts an -OH or -NH dipole
e.g. R-O: – – – H–N
2) The atom acts as a base if it uses the lone pair to capture H+
e.g. H+ + :NH2-R→ +NH3-R
3) The atom acts as a nucleophile when it shares the lone pair, i.e. bonds to another nucleus
E.g.,
In example 3, O is acting as a nucleophile because it has shared one of its lone pairs to bond to C, which is electron-deficient.
The curly arrow notation is commonly used to indicate movement of a pair of electrons, in this case from a non-bonded or lone pair position to form a new bond.
Hydrolysis is an attack by H2O, using O as a nucleophile, on a susceptible bond such as a peptide bond.

The bond is susceptible because the C atom of the C=O is electron deficient, as electrons are drawn towards the electronegative O atom of C=O. Because the C is electron deficient, it can accommodate the incoming electron pair (the maximum number of valence electrons on C, N or O is 8).
This sequence then produces a transition state, a semi-stable halfway stage of the reaction (Figure 4.8). The transition state gives rise to stable end products by breaking the C–N bond. This happens because the N atom can serve as a good leaving group, i.e. it can hold the electrons from the breaking bond as a lone pair.