Sam’s Health: Cystic Fibrosis
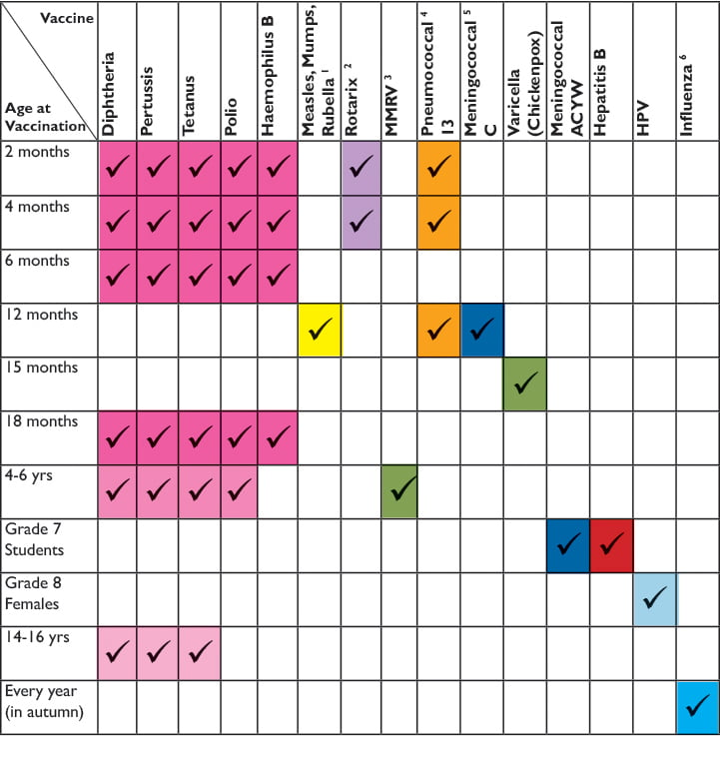
Sam is a happy, energetic child and fully up to date on his immunizations.
Some Concerns
Nancy & Paul were concerned about the following:
- They could hear Sam wheeze when he was breathing
- Greasy/oily stools in his diaper
Family physician referred Sam to a pediatrician.
Initial Visit
- Physical examination
- Blood work
- Chest x-ray
Physical Examination
Vital Signs
- Age: 2 years, 6 months
- Weight: 28 lbs. (lower 10%-tile)
- Height: 3 ft. 1 inch
- Pulse: 115 BPM
- Respirations: 30 breaths per minute
- Blood pressure: 95/60 mmHg
General Appearance
- Happy, energetic child
Head and Neck
- Runny nose but his ears are clear of fluid
- No enlarged lymph nodes in neck
Lungs
- Crackling sounds are present
- Coughing and wheezing are audible
Cardiovascular
- Normal
Abdominal
- No swelling present
Genitourinary
- Not assessed
Extremities
- Full mobility is present
- Pulse found in arms and legs
Neurological
- Normal reflexes
Follow-up appointment scheduled for the next day.
Lab & Diagnostic Results
- White blood cell count: values within normal limits
- WBC differential: lymphocytes, monocytes, eosinophils, & basophils are within normal limits. There is a slight elevation of neutrophils.
- Red blood cell count: values within normal limits
- Hematocrit: values within normal limits
- Platelet count: values within normal limits
- Sweat chloride test: 40 mmol/L
- Chest x-ray: some hyperinflation & bronchial wall thickening
Understanding Sweat Test Results
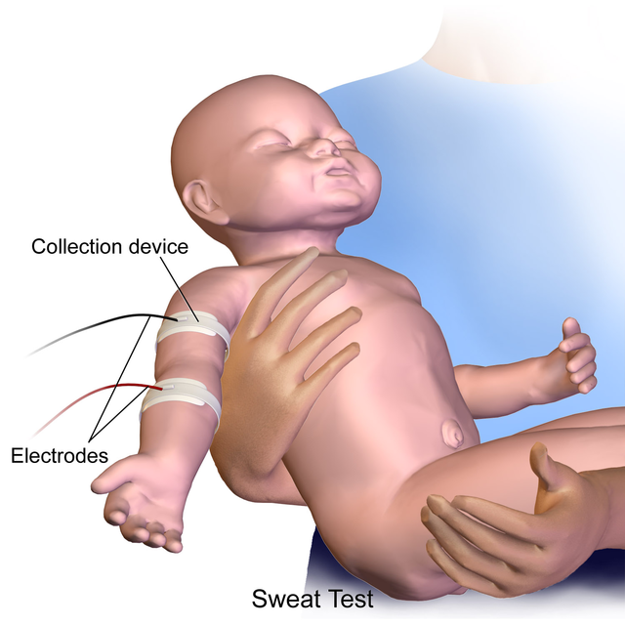
- People with CF have more chloride in their sweat
- Sweat chloride test will confirm the diagnosis
- Enough sweat is needed to do the test
- Full-term babies usually produce enough sweat by 2 weeks of age
- To understand what the sweat test results mean, a chloride level of:
- Less than or equal to 29 mmol/L = CF is unlikely regardless of age
- Between 30 – 59 mmol/L = CF is possible and additional testing is needed
- Greater than or equal to 60 mmol/L = CF is likely to be diagnosed
When sweat chloride test results fall between the range of 30-59 mmol/L, the sweat test is usually repeated.
Diagnosis
Cystic Fibrosis
- Life-threatening genetic disease
- Causes the body to create thick mucus
- Mucus builds up & obstructs ducts U tubes in the lungs, digestive track & pancreas
- May cause fatal infections & digestive issues
- Affects the sweat glands & male reproductive system
Newborn Screening
Newborn screening (NBS) for cystic fibrosis is done in the first few days after birth. By diagnosing CF early, CF health care providers can help parents learn ways to keep their child as healthy as possible and delay or prevent serious, lifelong health problems related to CF.
Research shows that children who receive CF care early in life have better nutrition and are healthier than those who are diagnosed later. Early diagnosis and treatment can:
- Improve growth
- Help keep lungs healthy
- Reduce hospital stays
- Add years to life
Timing
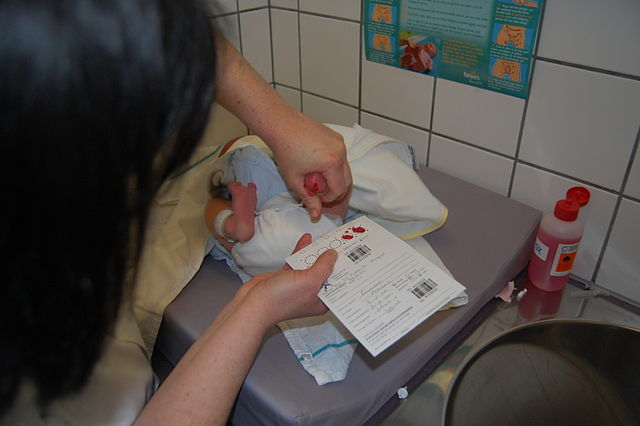 Newborn screening is done during the first few days of a baby’s life — usually by a health care provider in the hospital. A few drops of blood from a heel prick are placed on a special card, called a Guthrie card.
Newborn screening is done during the first few days of a baby’s life — usually by a health care provider in the hospital. A few drops of blood from a heel prick are placed on a special card, called a Guthrie card.
This card with the baby’s information is mailed to a special state laboratory that will test the blood sample for certain health conditions, including CF. In some states, newborn screening involves two blood samples, one at birth and one a few weeks later.
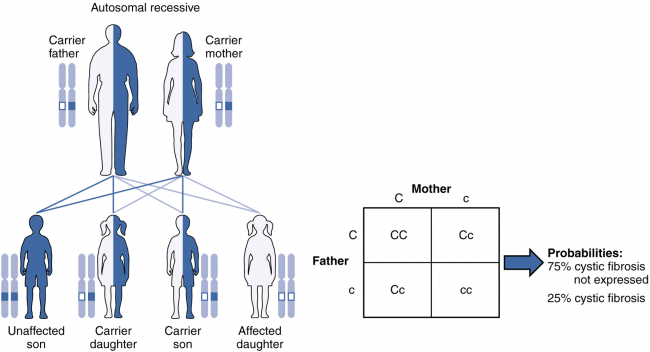
Genetics of Cystic Fibrosis
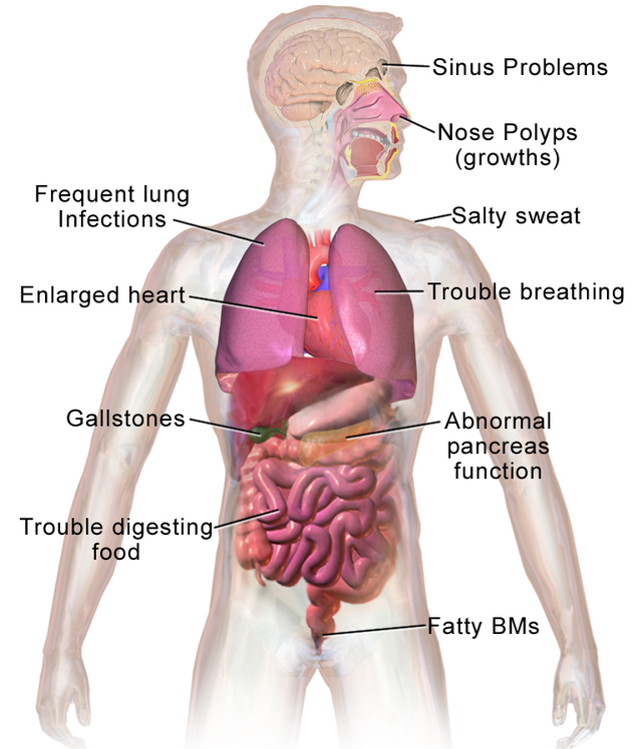
Symptoms
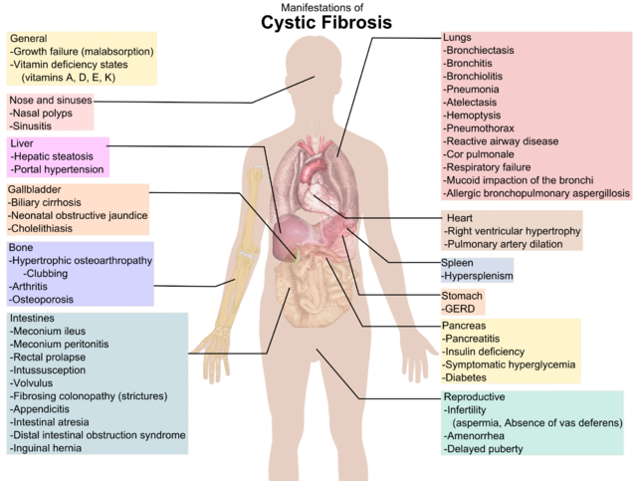
Complications
Respiratory System
- Recurrent attack of bronchitis & pneumonia
- Damage to the airways → bronchiectasis
- Nasal polyps
- Coughing of blood (hemoptysis)
- Pneumothorax
- Respiratory failure
Digestive System
- Malabsorption of nutrients
- Diabetes mellitus (pancreatic damage)
- Blockage of bile duct
- Intestinal obstruction
- Distal intestinal obstruction syndrome
Reproductive System
- Infertility
- Pregnancy may exacerbate the disease & cause complications
Other Systems
- Increased risk of osteoporosis
- Dehydration & electrolyte imbalances
- Clubbing of fingers & toes
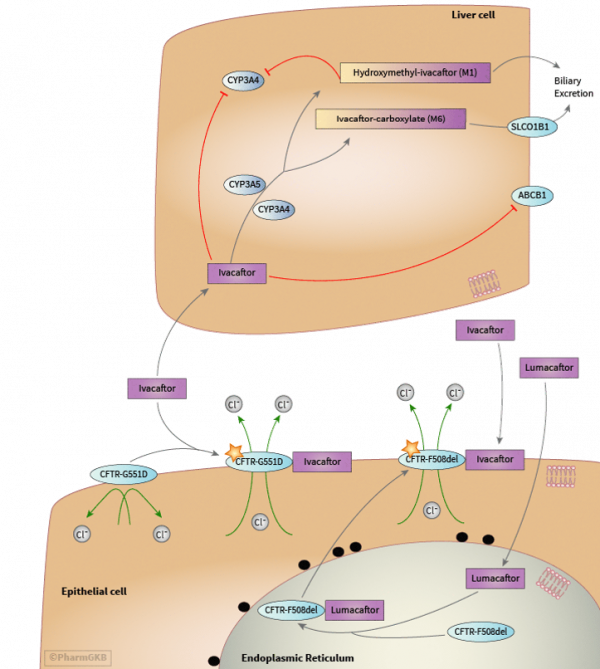
Treatment
At present there is no cure for CF. Treatment aims at alleviating the symptoms & reducing the incidence of complications. Careful follow-up & early & aggressive intervention is essential. It is advisable to obtain treatment at a centre that specializes in CF.
Aims of Treatment
- Reducing incidence & successful treatment of respiratory infections
- Providing sufficient nutrition
- Removal of & loosening mucus plugs in lung & airways
- Avoiding & treating intestinal obstruction
Chest Physical Therapy
- Loosens thick mucus & aids in expectoration
- Tapping the front & back of the chest with cupped hands
- May be done up to 4 times a day
- Patient can be sitting or lying on his abdomen
- Breathing exercises – coughing or huffing
- Aerobic exercise helps to loosen the mucus
- increases loss of salt & minerals
Nutritional Therapy
Nutritional therapy can take care of the malnutrition & vitamin deficiency that is common to CF patients. It involves:
- A well-balanced diet
- Intake of oral pancreatic enzymes to help in digestion & absorption
- Vitamin A, D, E, & K supplements
- High-calorie nutrient-filled shakes
- A high-salt diet or salt supplements (usually before exercise)
- A feeding tube may be used at night to give more calories
Medication
- Antibiotics – treating & preventing respiratory infections
- Mucolytics – aid in coughing out mucus, improves lung function
- Bronchodilators – keep airways patent & facilitate easier breathing
- Pancreatic enzyme supplements – improve absorption & digestion
- Vitamin supplements – including fat soluble vitamins
- New agents that improve chloride transport – IVACAFTOR
| Disease Modifying Drug | |
| Kalydeco G511D | (ivacaftor) a pill for people ages 6 and older who have the G551D mutation of CF which helps the defective CFTR protein work at the surface of the cell |
| Symptom Management | |
| PULMOZYME (Dornase Alfa) | is an inhaled medication used to help thin the mucus |
| Hypertonic Saline | to draw more water into the airways and make it easier to cough out the mucus. |
| Ibuprofen | anti-inflammatory to slow the rate of lung function decline |
| CREON | pancreatic enzymes to help people with CF digest their food |
| COTAZYM | pancreatic enzymes to help people with CF digest their food |
| FLOVENT | inhaled steroid treatment Open benefit |
| ADVAIR | inhaled steroid treatment for asthma |
| Salbutamol (MDI or Nebules) | bronchodilator |
| Infection Control | |
| CAYSTON | (aztreonam) inhaled antibiotic for the treatment of CF |
| Zithromax (Azithromycin) | is a commonly used antibiotic to treat pneumonia |
| COLY-MYCIN | antibiotic |
| TOBI Podhaler | (tobramycin) inhaled antibiotic with Podhaler device to help fight the germ Pseudomonas aeruginosa |
| TOBI Inhaled Tobramycin 300mg | (tobramycin) inhaled antibiotic to help fight the germ Pseudomonas aeruginosa |
Surgical Treatment
- Removal of nasal polyps
- Removal of mucus obstructing airways (endoscope)
- Oxygen therapy
- Alimentation through feeding tube
- Relieving intestinal obstruction
- Lung transplant
Caregiver Suggestions
- CF symptoms may worsen over time
- Managing CF takes a team approach
- Staying organized makes treatment easier
- Preventing infection is essential
- Focusing on nutrition is a must
- Exercise can keep everyone healthy
- Emotional distress is real
- Your loved one may need a lung transplant someday
- It’s important to take steps to help your child transition to adulthood
- You need to make time to take care of yourself, too
