1.9 Nervous System Infections
Bacterial Diseases of the Nervous System
By the end of this section, you will be able to:
- Identify the most common bacteria that can cause infections of the nervous system
- Compare the major characteristics of specific bacterial diseases affecting the nervous system
Bacterial infections that affect the nervous system are serious and can be life-threatening. Fortunately, there are only a few bacterial species commonly associated with neurological infections.
Bacterial Meningitis
Bacterial meningitis is one of the most serious forms of meningitis. Bacteria that cause meningitis often gain access to the CNS through the bloodstream after trauma or as a result of the action of bacterial toxins. Bacteria may also spread from structures in the upper respiratory tract, such as the oropharynx, nasopharynx, sinuses, and middle ear. Patients with head wounds or cochlear implants (an electronic device placed in the inner ear) are also at risk for developing meningitis.
Many of the bacteria that can cause meningitis are commonly found in healthy people. The most common causes of non-neonatal bacterial meningitis are Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae. All three of these bacterial pathogens are spread from person to person by respiratory secretions. Each can colonize and cross through the mucous membranes of the oropharynx and nasopharynx, and enter the blood. Once in the blood, these pathogens can disseminate throughout the body and are capable of both establishing an infection and triggering inflammation in any body site, including the meninges (Figure 1.). Without appropriate systemic antibacterial therapy, the case-fatality rate can be as high as 70%, and 20% of those survivors may be left with irreversible nerve damage or tissue destruction, resulting in hearing loss, neurologic disability, or loss of a limb. Mortality rates are much lower (as low as 15%) in populations where appropriate therapeutic drugs and preventive vaccines are available.
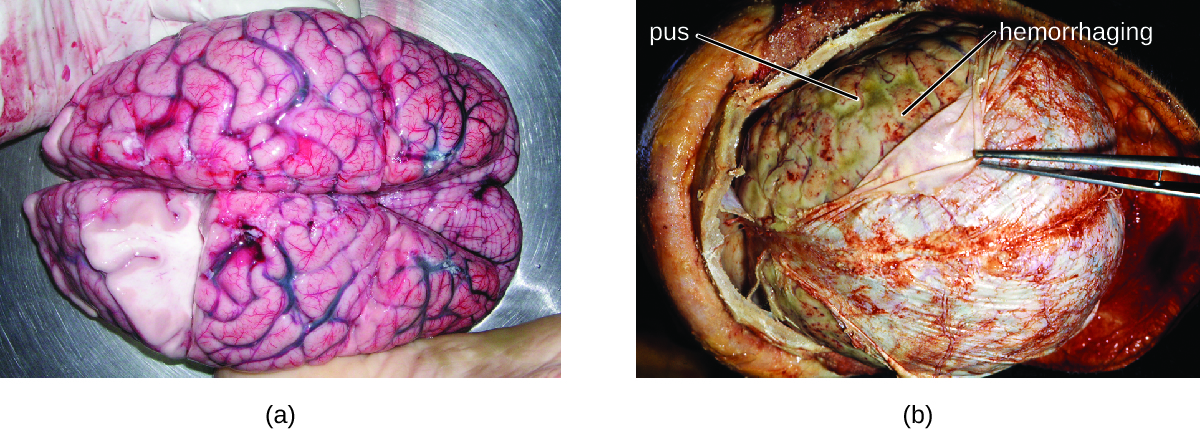
A variety of other bacteria, including Listeria monocytogenes and Escherichia coli, are also capable of causing meningitis. These bacteria cause infections of the arachnoid mater and CSF after spreading through the circulation in blood or by spreading from an infection of the sinuses or nasopharynx. Streptococcus agalactiae, commonly found in the microbiota of the vagina and gastrointestinal tract, can also cause bacterial meningitis in newborns after transmission from the mother either before or during birth.
The profound inflammation caused by these microbes can result in early symptoms that include severe headache, fever, confusion, nausea, vomiting, photophobia, and stiff neck. Systemic inflammatory responses associated with some types of bacterial meningitis can lead to hemorrhaging and purpuric lesions on the skin, followed by even more severe conditions that include shock, convulsions, coma, and death—in some cases, in the span of just a few hours.
Diagnosis of bacterial meningitis is best confirmed by the analysis of CSF obtained by a lumbar puncture. Abnormal levels of polymorphonuclear neutrophils (PMNs) (> 10 PMNs/mm3), glucose (< 45 mg/dL), and protein (> 45 mg/dL) in the CSF are suggestive of bacterial meningitis. Characteristics of specific forms of bacterial meningitis are detailed in the subsections that follow.
Meningococcal Meningitis
Meningococcal meningitis is a serious infection caused by the gram-negative coccus N. meningitidis. In some cases, death can occur within a few hours of the onset of symptoms. Nonfatal cases can result in irreversible nerve damage, resulting in hearing loss and brain damage, or amputation of extremities because of tissue necrosis.
Meningococcal meningitis can infect people of any age, but its prevalence is highest among infants, adolescents, and young adults. Meningococcal meningitis was once the most common cause of meningitis epidemics in human populations. This is still the case in a swath of sub-Saharan Africa known as the meningitis belt, but meningococcal meningitis epidemics have become rare in most other regions, thanks to meningococcal vaccines. However, outbreaks can still occur in communities, schools, colleges, prisons, and other populations where people are in close direct contact.
N. meningitidis has a high affinity for mucosal membranes in the oropharynx and nasopharynx. Contact with respiratory secretions containing N. meningitidis is an effective mode of transmission. The pathogenicity of N. meningitidis is enhanced by virulence factors that contribute to the rapid progression of the disease. These include lipooligosaccharide (LOS) endotoxin, type IV pili for attachment to host tissues, and polysaccharide capsules that help the cells avoid phagocytosis and complement-mediated killing. Additional virulence factors include IgA protease (which breaks down IgA antibodies), the invasion factors Opa, Opc, and porin (which facilitate transcellular entry through the blood-brain barrier), iron-uptake factors (which strip heme units from hemoglobin in host cells and use them for growth), and stress proteins that protect bacteria from reactive oxygen molecules.
A unique sign of meningococcal meningitis is the formation of a petechial rash on the skin or mucous membranes, characterized by tiny, red, flat, hemorrhagic lesions. This rash, which appears soon after disease onset, is a response to LOS endotoxin and adherence virulence factors that disrupt the endothelial cells of capillaries and small veins in the skin. The blood vessel disruption triggers the formation of tiny blood clots, causing blood to leak into the surrounding tissue. As the infection progresses, the levels of virulence factors increase, and the hemorrhagic lesions can increase in size as blood continues to leak into tissues. Lesions larger than 1.0 cm usually occur in patients developing shock, as virulence factors cause increased hemorrhage and clot formation. Sepsis, as a result of systemic damage from meningococcal virulence factors, can lead to rapid multiple organ failure, shock, disseminated intravascular coagulation, and death.
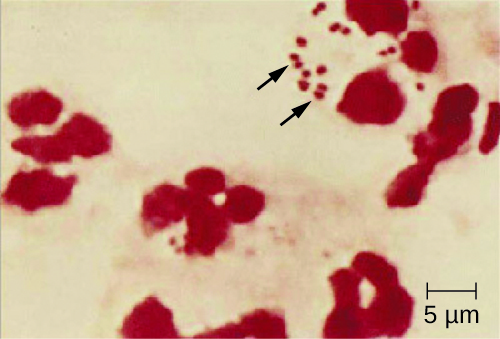
Because meningococcoal meningitis progresses so rapidly, a greater variety of clinical specimens are required for the timely detection of N. meningitidis. Required specimens can include blood, CSF, naso- and oropharyngeal swabs, urethral and endocervical swabs, petechial aspirates, and biopsies. Safety protocols for handling and transport of specimens suspected of containing N. meningitidis should always be followed, since cases of fatal meningococcal disease have occurred in healthcare workers exposed to droplets or aerosols from patient specimens. A prompt presumptive diagnosis of meningococcal meningitis can occur when CSF is directly evaluated by Gram stain, revealing extra- and intracellular gram-negative diplococci with a distinctive coffee-bean microscopic morphology associated with PMNs (Figure 2.). Identification can also be made directly from CSF using latex agglutination and immunochromatographic rapid diagnostic tests specific for N. meningitidis. Species identification can also be performed using DNA sequence-based typing schemes for hypervariable outer membrane proteins of N. meningitidis, which has replaced sero(sub)typing.
Meningococcal infections can be treated with antibiotic therapy, and third-generation cephalosporins are most often employed. However, because outcomes can be negative even with treatment, preventive vaccination is the best form of treatment. In 2010, countries in Africa’s meningitis belt began using a new serogroup A meningococcal conjugate vaccine. This program has dramatically reduced the number of cases of meningococcal meningitis by conferring individual and herd immunity.
Twelve different capsular serotypes of N. meningitidis are known to exist. Serotypes A, B, C, W, X, and Y are the most prevalent worldwide. The CDC recommends that children between 11–12 years of age be vaccinated with a single dose of a quadrivalent vaccine that protects against serotypes A, C, W, and Y, with a booster at age 16. An additional booster or injections of serogroup B meningococcal vaccine may be given to individuals in high-risk settings (such as epidemic outbreaks on college campuses).
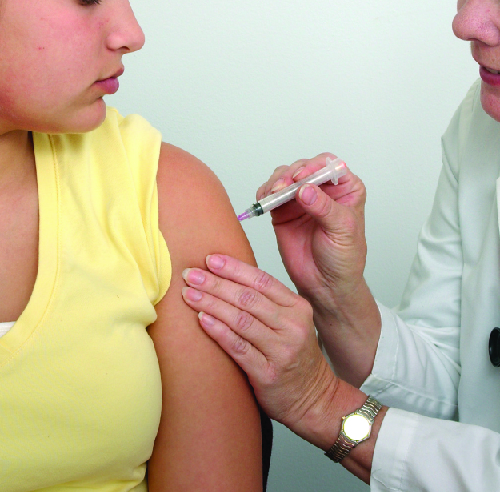
College students living in dorms or communal housing are at increased risk for contracting epidemic meningitis. From 2011 to 2015, there have been at least nine meningococcal outbreaks on college campuses in the United States. These incidents involved a total of 43 students (of whom four died). In spite of rapid diagnosis and aggressive antimicrobial treatment, several of the survivors suffered from amputations or serious neurological problems.
Prophylactic vaccination of first-year college students living in dorms is recommended by the CDC, and insurance companies now cover meningococcal vaccination for students in college dorms. Some colleges have mandated vaccination with meningococcal conjugate vaccine for certain students entering college (Figure 3.).
Pneumococcal Meningitis
Pneumococcal meningitis is caused by the encapsulated gram-positive bacterium S. pneumoniae(pneumococcus, also called strep pneumo). This organism is commonly found in the microbiota of the pharynx of 30–70% of young children, depending on the sampling method, while S. pneumoniae can be found in fewer than 5% of healthy adults. Although it is often present without disease symptoms, this microbe can cross the blood-brain barrier in susceptible individuals. In some cases, it may also result in septicemia. Since the introduction of the Hib vaccine, S. pneumoniae has become the leading cause of meningitis in humans aged 2 months through adulthood.
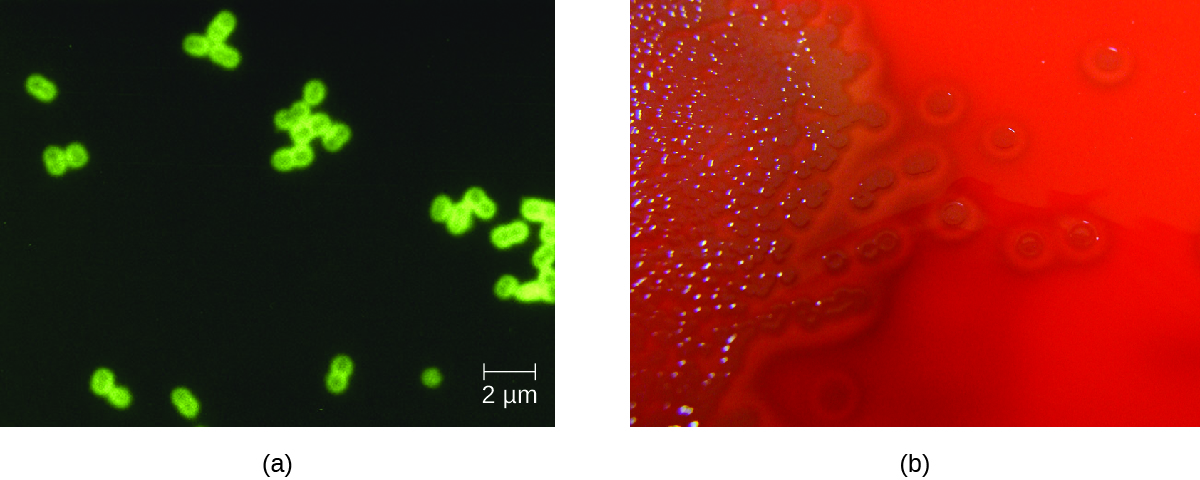
S. pneumoniae can be identified in CSF samples using gram-stained specimens, latex agglutination, and immunochromatographic RDT specific for S. pneumoniae. In gram-stained samples, S. pneumoniae appears as gram-positive, lancet-shaped diplococci (Figure 4.). Identification of S. pneumoniae can also be achieved using cultures of CSF and blood, and at least 93 distinct serotypes can be identified based on the quellung reaction to unique capsular polysaccharides. PCR and RT-PCR assays are also available to confirm identification.
Major virulence factors produced by S. pneumoniae include PI-1 pilin for adherence to host cells (pneumococcal adherence) and virulence factor B (PavB) for attachment to cells of the respiratory tract; choline-binding proteins (cbpA) that bind to epithelial cells and interfere with immune factors IgA and C3; and the cytoplasmic bacterial toxin pneumolysin that triggers an inflammatory response.
With the emergence of drug-resistant strains of S. pneumoniae, pneumococcal meningitis is typically treated with broad-spectrum antibiotics, such as levofloxacin, cefotaxime, penicillin, or other β-lactam antibiotics.
Haemophilus influenzae Type b
Meningitis due to H. influenzae serotype b (Hib), an encapsulated pleomorphic gram-negative coccobacilli, is now uncommon in most countries, because of the use of the effective Hib vaccine. Without the use of the Hib vaccine, H. influenzae can be the primary cause of meningitis in children 2 months thru 5 years of age. H. influenzae can be found in the throats of healthy individuals, including infants and young children. By five years of age, most children have developed immunity to this microbe. Infants older than 2 months of age, however, do not produce a sufficient protective antibody response and are susceptible to serious disease. The intracranial pressure caused by this infection leads to a 5% mortality rate and 20% incidence of deafness or brain damage in survivors.
H. influenzae produces at least 16 different virulence factors, including LOS, which triggers inflammation, and Haemophilus adhesion and penetration factor (Hap), which aids in attachment and invasion into respiratory epithelial cells. The bacterium also has a polysaccharide capsule that helps it avoid phagocytosis, as well as factors such as IgA1 protease and P2 protein that allow it to evade antibodies secreted from mucous membranes. In addition, factors such as hemoglobin-binding protein (Hgp) and transferrin-binding protein (Tbp) acquire iron from hemoglobin and transferrin, respectively, for bacterial growth.
Preliminary diagnosis of H. influenzae infections can be made by direct PCR and a smear of CSF. Stained smears will reveal intracellular and extracellular PMNs with small, pleomorphic, gram-negative coccobacilli or filamentous forms that are characteristic of H. influenzae. Initial confirmation of this genus can be based on its fastidious growth on chocolate agar. Identification is confirmed with requirements for exogenous biochemical growth cofactors NAD and heme (by MALDI-TOF), latex agglutination, and RT-PCR.
Meningitis caused by H. influenzae is usually treated with doxycycline, fluoroquinolones, second- and third-generation cephalosporins, and carbapenems. The best means of preventing H. influenza infection is with the use of the Hib polysaccharide conjugate vaccine. It is recommended that all children receive this vaccine at 2, 4, and 6 months of age, with a final booster dose at 12 to 15 months of age.
Neonatal Meningitis
S. agalactiae, Group B streptococcus (GBS), is an encapsulated gram-positive bacterium that is the most common cause of neonatal meningitis, a term that refers to meningitis occurring in babies up to 3 months of age. S. agalactiae can also cause meningitis in people of all ages and can be found in the urogenital and gastrointestinal microbiota of about 10–30% of humans.
Neonatal infection occurs as either early onset or late-onset disease. Early onset disease is defined as occurring in infants up to 7 days old. The infant initially becomes infected by S. agalactiae during childbirth, when the bacteria may be transferred from the mother’s vagina. Incidence of early onset neonatal meningitis can be greatly reduced by giving intravenous antibiotics to the mother during labor.
Late-onset neonatal meningitis occurs in infants between 1 week and 3 months of age. Infants born to mothers with S. agalactiae in the urogenital tract have a higher risk of late-onset menigitis, but late-onset infections can be transmitted from sources other than the mother; often, the source of infection is unknown. Infants who are born prematurely (before 37 weeks of pregnancy) or to mothers who develop a fever also have a greater risk of contracting late-onset neonatal meningitis.
Signs and symptoms of early onset disease include temperature instability, apnea (cessation of breathing), bradycardia (slow heart rate), hypotension, difficulty feeding, irritability, and limpness. When asleep, the baby may be difficult to wake up. Symptoms of late-onset disease are more likely to include seizures, bulging fontanel (soft spot), stiff neck, hemiparesis (weakness on one side of the body), and opisthotonos (rigid body with arched back and head thrown backward).
S. agalactiae produces at least 12 virulence factors that include FbsA that attaches to host cell surface proteins, PI-1 pili that promotes the invasion of human endothelial cells, a polysaccharide capsule that prevents the activation of the alternative complement pathway and inhibits phagocytosis, and the toxin CAMP factor, which forms pores in host cell membranes and binds to IgG and IgM antibodies.
Diagnosis of neonatal meningitis is often, but not uniformly, confirmed by positive results from cultures of CSF or blood. Tests include routine culture, antigen detection by enzyme immunoassay, serotyping of different capsule types, PCR, and RT-PCR. It is typically treated with β-lactam antibiotics such as intravenous penicillin or ampicillin plus gentamicin. Even with treatment, roughly 10% mortality is seen in infected neonates.
- Which groups are most vulnerable to each of the bacterial meningitis diseases?
- For which of the bacterial meningitis diseases are there vaccines presently available?
- Which organism can cause epidemic meningitis?
Clostridium-Associated Diseases
Species in the genus Clostridium are gram-positive, endospore-forming rods that are obligate anaerobes. Endospores of Clostridium spp. are widespread in nature, commonly found in soil, water, feces, sewage, and marine sediments. Clostridium spp. produce more types of protein exotoxins than any other bacterial genus, including two exotoxins with protease activity that are the most potent known biological toxins: botulinum neurotoxin (BoNT) and tetanus neurotoxin (TeNT). These two toxins have lethal doses of 0.2–10 ng per kg body weight.
BoNT can be produced by unique strains of C. butyricum, and C. baratii; however, it is primarily associated with C. botulinum and the condition of botulism. TeNT, which causes tetanus, is only produced by C. tetani. These powerful neural exotoxins are the primary virulence factors for these pathogens.
Diagnosis of tetanus or botulism typically involves bioassays that detect the presence of BoNT and TeNT in fecal specimens, blood (serum), or suspect foods. In addition, both C. botulinum and C. tetani can be isolated and cultured using commercially available media for anaerobes. ELISA and RT-PCR tests are also available.
Tetanus
Tetanus is a noncommunicable disease characterized by uncontrollable muscle spasms (contractions) caused by the action of TeNT. It generally occurs when C. tetani infects a wound and produces TeNT, which rapidly binds to neural tissue, resulting in an intoxication (poisoning) of neurons. Depending on the site and extent of infection, cases of tetanus can be described as localized, cephalic, or generalized. Generalized tetanus that occurs in a newborn is called neonatal tetanus.
Localized tetanus occurs when TeNT only affects the muscle groups close to the injury site. There is no CNS involvement, and the symptoms are usually mild, with localized muscle spasms caused by a dysfunction in the surrounding neurons. Individuals with partial immunity—especially previously vaccinated individuals who neglect to get the recommended booster shots—are most likely to develop localized tetanus as a result of C. tetani infecting a puncture wound.
Cephalic tetanus is a rare, localized form of tetanus generally associated with wounds on the head or face. In rare cases, it has occurred in cases of otitis media (middle ear infection). Cephalic tetanus often results in patients seeing double images, because of the spasms affecting the muscles that control eye movement.
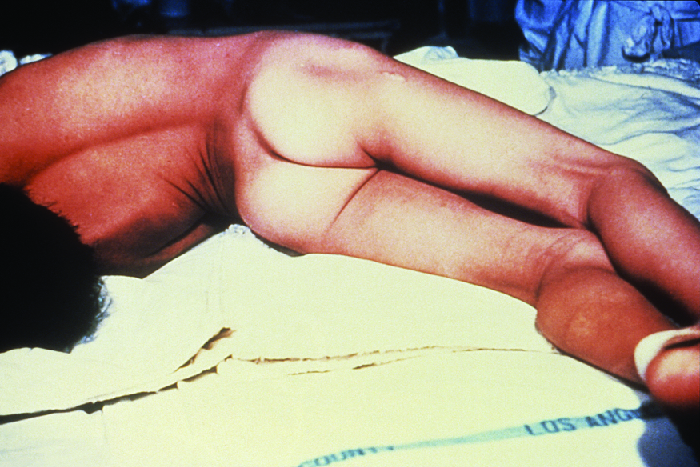
Both localized and cephalic tetanus may progress to generalized tetanus—a much more serious condition—if TeNT is able to spread further into body tissues. In generalized tetanus, TeNT enters neurons of the PNS. From there, TeNT travels from the site of the wound, usually on an extremity of the body, retrograde (back up) to inhibitory neurons in the CNS. There, it prevents the release of gamma aminobutyric acid (GABA), the neurotransmitter responsible for muscle relaxation. The resulting muscle spasms often first occur in the jaw muscles, leading to the characteristic symptom of lockjaw (inability to open the mouth). As the toxin progressively continues to block neurotransmitter release, other muscles become involved, resulting in uncontrollable, sudden muscle spasms that are powerful enough to cause tendons to rupture and bones to fracture. Spasms in the muscles in the neck, back, and legs may cause the body to form a rigid, stiff arch, a posture called opisthotonos (Figure 5.). Spasms in the larynx, diaphragm, and muscles of the chest restrict the patient’s ability to swallow and breathe, eventually leading to death by asphyxiation (insufficient supply of oxygen).
Neonatal tetanus typically occurs when the stump of the umbilical cord is contaminated with spores of C. tetani after delivery. Although this condition is rare in the United States, neonatal tetanus is a major cause of infant mortality in countries that lack maternal immunization for tetanus and where birth often occurs in unsanitary conditions. At the end of the first week of life, infected infants become irritable, feed poorly, and develop rigidity with spasms. Neonatal tetanus has a very poor prognosis with a mortality rate of 70%–100%.
Treatment for patients with tetanus includes assisted breathing through the use of a ventilator, wound debridement, fluid balance, and antibiotic therapy with metronidazole or penicillin to halt the growth of C. tetani. In addition, patients are treated with TeNT antitoxin, preferably in the form of human immunoglobulin to neutralize nonfixed toxin and benzodiazepines to enhance the effect of GABA for muscle relaxation and anxiety.
A tetanus toxoid (TT) vaccine is available for protection and prevention of tetanus. It is the T component of vaccines such as DTaP, Tdap, and Td. The CDC recommends children receive doses of the DTaP vaccine at 2, 4, 6, and 15–18 months of age and another at 4–6 years of age. One dose of Td is recommended for adolescents and adults as a TT booster every 10 years.
Botulism
Botulism is a rare but frequently fatal illness caused by intoxication by BoNT. It can occur either as the result of an infection by C. botulinum, in which case the bacteria produce BoNT in vivo, or as the result of a direct introduction of BoNT into tissues.
Infection and production of BoNT in vivo can result in wound botulism, infant botulism, and adult intestinal toxemia. Wound botulism typically occurs when C. botulinum is introduced directly into a wound after a traumatic injury, deep puncture wound, or injection site. Infant botulism, which occurs in infants younger than 1 year of age, and adult intestinal toxemia, which occurs in immunocompromised adults, results from ingesting C. botulinum endospores in food. The endospores germinate in the body, resulting in the production of BoNT in the intestinal tract.
Intoxications occur when BoNT is produced outside the body and then introduced directly into the body through food (foodborne botulism), air (inhalation botulism), or a clinical procedure (iatrogenic botulism). Foodborne botulism, the most common of these forms, occurs when BoNT is produced in contaminated food and then ingested along with the food. Inhalation botulism is rare because BoNT is unstable as an aerosol and does not occur in nature; however, it can be produced in the laboratory and was used (unsuccessfully) as a bioweapon by terrorists in Japan in the 1990s. A few cases of accidental inhalation botulism have also occurred. Iatrogenic botulism is also rare; it is associated with injections of BoNT used for cosmetic purposes.
When BoNT enters the bloodstream in the gastrointestinal tract, wound, or lungs, it is transferred to the neuromuscular junctions of motor neurons where it binds irreversibly to presynaptic membranes and prevents the release of acetylcholine from the presynaptic terminal of motor neurons into the neuromuscular junction. The consequence of preventing acetylcholine release is the loss of muscle activity, leading to muscle relaxation and eventually paralysis.
If BoNT is absorbed through the gastrointestinal tract, early symptoms of botulism include blurred vision, drooping eyelids, difficulty swallowing, abdominal cramps, nausea, vomiting, constipation, or possibly diarrhea. This is followed by progressive flaccid paralysis, a gradual weakening and loss of control over the muscles. A patient’s experience can be particularly terrifying, because hearing remains normal, consciousness is not lost, and he or she is fully aware of the progression of his or her condition. In infants, notable signs of botulism include weak cry, decreased ability to suckle, and hypotonia (limpness of head or body). Eventually, botulism ends in death from respiratory failure caused by the progressive paralysis of the muscles of the upper airway, diaphragm, and chest.
Botulism is treated with an antitoxin specific for BoNT. If administered in time, the antitoxin stops the progression of paralysis but does not reverse it. Once the antitoxin has been administered, the patient will slowly regain neurological function, but this may take several weeks or months, depending on the severity of the case. During recovery, patients generally must remain hospitalized and receive breathing assistance through a ventilator.
- How frequently should the tetanus vaccination be updated in adults?
- What are the most common causes of botulism?
- Why is botulism not treated with an antibiotic?
Although it is the most toxic biological material known to man, botulinum toxin is often intentionally injected into people to treat other conditions. Type A botulinum toxin is used cosmetically to reduce wrinkles. The injection of minute quantities of this toxin into the face causes the relaxation of facial muscles, thereby giving the skin a smoother appearance. Eyelid twitching and crossed eyes can also be treated with botulinum toxin injections. Other uses of this toxin include the treatment of hyperhidrosis (excessive sweating). In fact, botulinum toxin can be used to moderate the effects of several other apparently nonmicrobial diseases involving inappropriate nerve function. Such diseases include cerebral palsy, multiple sclerosis, and Parkinson’s disease. Each of these diseases is characterized by a loss of control over muscle contractions; treatment with botulinum toxin serves to relax contracted muscles.
Listeriosis
Listeria monocytogenes is a nonencapsulated, nonsporulating, gram-positive rod and a foodborne pathogen that causes listeriosis. At-risk groups include pregnant women, neonates, the elderly, and the immunocompromised. Listeriosis leads to meningitis in about 20% of cases, particularly neonates and patients over the age of 60. The CDC identifies listeriosis as the third leading cause of death due to foodborne illness, with overall mortality rates reaching 16%. In pregnant women, listeriosis can cause also cause spontaneous abortion in pregnant women because of the pathogen’s unique ability to cross the placenta.
L. monocytogenes is generally introduced into food items by contamination with soil or animal manure used as fertilizer. Foods commonly associated with listeriosis include fresh fruits and vegetables, frozen vegetables, processed meats, soft cheeses, and raw milk. Unlike most other foodborne pathogens, Listeria is able to grow at temperatures between 0 °C and 50 °C, and can therefore continue to grow, even in refrigerated foods.
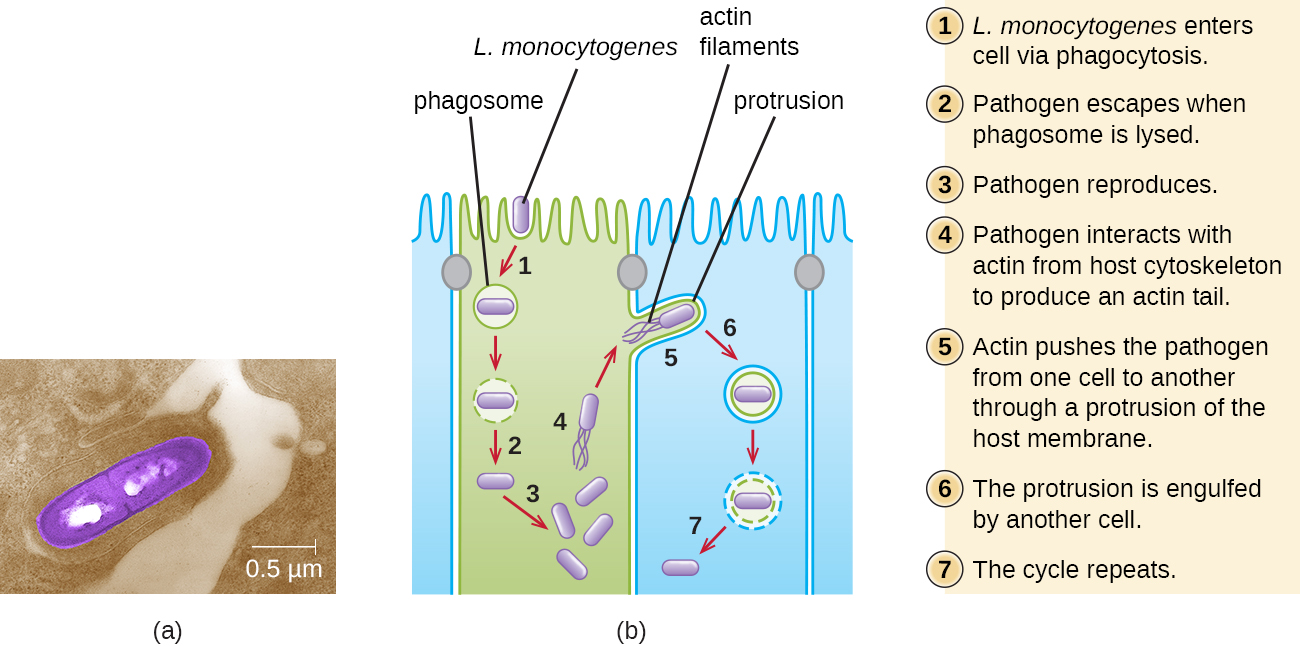
Ingestion of contaminated food leads initially to infection of the gastrointestinal tract. However, L. monocytogenes produces several unique virulence factors that allow it to cross the intestinal barrier and spread to other body systems. Surface proteins called internalins (InlA and InlB) help L. monocytogenes invade nonphagocytic cells and tissues, penetrating the intestinal wall and becoming disseminating through the circulatory and lymphatic systems. Internalins also enable L. monocytogenes to breach other important barriers, including the blood-brain barrier and the placenta. Within tissues, L. monocytogenes uses other proteins called listeriolysin O and ActA to facilitate intercellular movement, allowing the infection to spread from cell to cell (Figure 6.).
L. monocytogenes is usually identified by cultivation of samples from a normally sterile site (e.g., blood or CSF). Recovery of viable organisms can be enhanced using cold enrichment by incubating samples in a broth at 4 °C for a week or more. Distinguishing types and subtypes of L. monocytogenes—an important step for diagnosis and epidemiology—is typically done using pulsed-field gel electrophoresis. Identification can also be achieved using chemiluminescence DNA probe assays and MALDI-TOF.
Treatment for listeriosis involves antibiotic therapy, most commonly with ampicillin and gentamicin. There is no vaccine available.
- How does Listeria enter the nervous system?
Hansen’s Disease (Leprosy)
Hansen’s disease (also known as leprosy) is caused by a long, thin, filamentous rod-shaped bacterium Mycobacterium leprae, an obligate intracellular pathogen. M. leprae is classified as gram-positive bacteria, but it is best visualized microscopically with an acid-fast stain and is generally referred to as an acid-fast bacterium. Hansen’s disease affects the PNS, leading to permanent damage and loss of appendages or other body parts.
Hansen’s disease is communicable but not highly contagious; approximately 95% of the human population cannot be easily infected because they have a natural immunity to M. leprae. Person-to-person transmission occurs by inhalation into nasal mucosa or prolonged and repeated contact with infected skin. Armadillos, one of only five mammals susceptible to Hansen’s disease, have also been implicated in transmission of some cases.
In the human body, M. leprae grows best at the cooler temperatures found in peripheral tissues like the nose, toes, fingers, and ears. Some of the virulence factors that contribute to M. leprae’s pathogenicity are located on the capsule and cell wall of the bacterium. These virulence factors enable it to bind to and invade Schwann cells, resulting in progressive demyelination that gradually destroys neurons of the PNS. The loss of neuronal function leads to hypoesthesia (numbness) in infected lesions. M. leprae is readily phagocytized by macrophages but is able to survive within macrophages in part by neutralizing reactive oxygen species produced in the oxidative burst of the phagolysosome. Like L. monocytogenes, M. leprae also can move directly between macrophages to avoid clearance by immune factors.
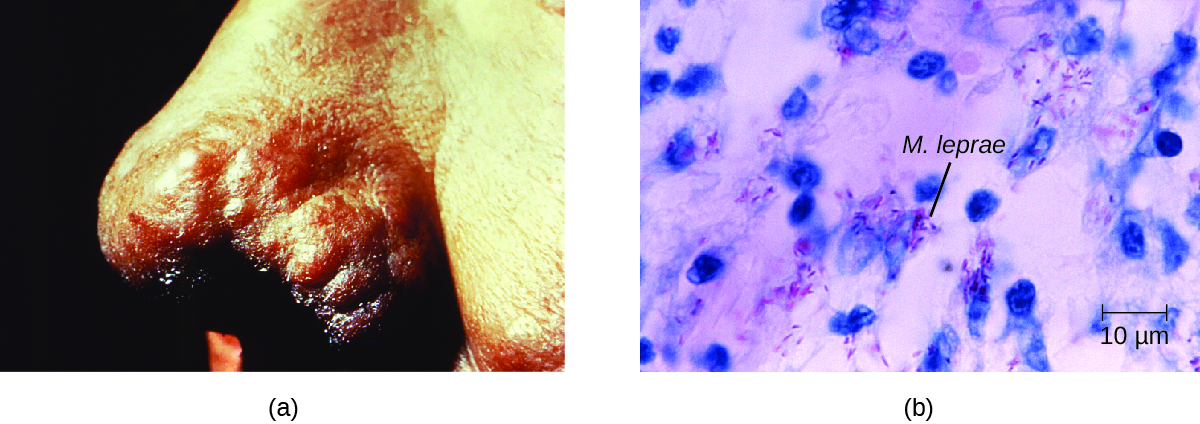
The extent of the disease is related to the immune response of the patient. Initial symptoms may not appear for as long as 2 to 5 years after infection. These often begin with small, blanched, numb areas of the skin. In most individuals, these will resolve spontaneously, but some cases may progress to a more serious form of the disease. Tuberculoid (paucibacillary) Hansen’s disease is marked by the presence of relatively few (three or less) flat, blanched skin lesions with small nodules at the edges and few bacteria present in the lesion. Although these lesions can persist for years or decades, the bacteria are held in check by an effective immune response including cell-mediated cytotoxicity. Individuals who are unable to contain the infection may later develop lepromatous (multibacillary) Hansen’s disease. This is a progressive form of the disease characterized by nodules filled with acid-fast bacilli and macrophages. Impaired function of infected Schwann cells leads to peripheral nerve damage, resulting in sensory loss that leads to ulcers, deformities, and fractures. Damage to the ulnar nerve (in the wrist) by M. leprae is one of the most common causes of crippling of the hand. In some cases, chronic tissue damage can ultimately lead to loss of fingers or toes. When mucosal tissues are also involved, disfiguring lesions of the nose and face can also occur (Figure 7.).
Hansen’s disease is diagnosed on the basis of clinical signs and symptoms of the disease, and confirmed by the presence of acid-fast bacilli on skin smears or in skin biopsy specimens (Figure 7.). M. leprae does not grow in vitro on any known laboratory media, but it can be identified by culturing in vivo in the footpads of laboratory mice or armadillos. Where needed, PCR and genotyping of M. leprae DNA in infected human tissue may be performed for diagnosis and epidemiology.
Hansen’s disease responds well to treatment and, if diagnosed and treated early, does not cause disability. In the United States, most patients with Hansen’s disease are treated in ambulatory care clinics in major cities by the National Hansen’s Disease program, the only institution in the United States exclusively devoted to Hansen’s disease. Since 1995, WHO has made multidrug therapy for Hansen’s disease available free of charge to all patients worldwide. As a result, global prevalence of Hansen’s disease has declined from about 5.2 million cases in 1985 to roughly 176,000 in 2014. Multidrug therapy consists of dapsone and rifampicin for all patients and a third drug, clofazimin, for patients with multibacillary disease.
Currently, there is no universally accepted vaccine for Hansen’s disease. India and Brazil use a tuberculosis vaccine against Hansen’s disease because both diseases are caused by species of Mycobacterium. The effectiveness of this method is questionable, however, since it appears that the vaccine works in some populations but not in others.
- What prevents the progression from tuberculoid to lepromatus leprosy?
- Why does Hansen’s disease typically affect the nerves of the extremities?
Disfiguring, deadly diseases like leprosy have historically been stigmatized in many cultures. Before leprosy was understood, victims were often isolated in leper colonies, a practice mentioned frequently in ancient texts, including the Bible. But leper colonies are not just an artifact of the ancient world. In Hawaii, a leper colony established in the late nineteenth century persisted until the mid-twentieth century, its residents forced to live in deplorable conditions. Although leprosy is a communicable disease, it is not considered contagious (easily communicable), and it certainly does not pose enough of a threat to justify the permanent isolation of its victims. Today, we reserve the practices of isolation and quarantine to patients with more dangerous diseases, such as Ebola or multiple-drug-resistant bacteria like Mycobacterium tuberculosis and Staphylococcus aureus. The ethical argument for this practice is that isolating infected patients is necessary to prevent the transmission and spread of highly contagious diseases—even when it goes against the wishes of the patient.
Of course, it is much easier to justify the practice of temporary, clinical quarantining than permanent social segregation, as occurred in leper colonies. In the 1980s, there were calls by some groups to establish camps for people infected with AIDS. Although this idea was never actually implemented, it begs the question—where do we draw the line? Are permanent isolation camps or colonies ever medically or socially justifiable? Suppose there were an outbreak of a fatal, contagious disease for which there is no treatment. Would it be justifiable to impose social isolation on those afflicted with the disease? How would we balance the rights of the infected with the risk they pose to others? To what extent should society expect individuals to put their own health at risk for the sake of treating others humanely?
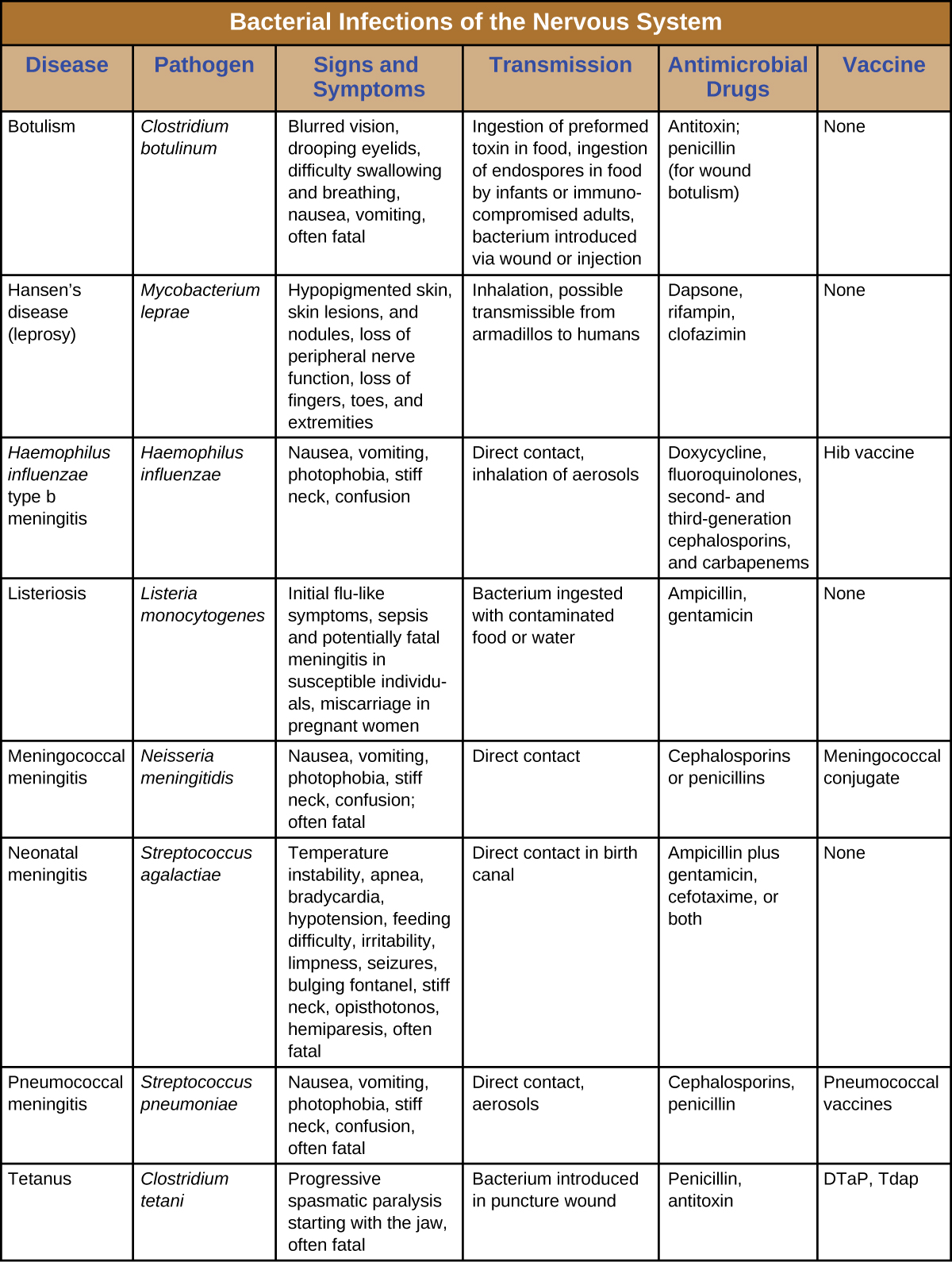
Despite the formidable defenses protecting the nervous system, a number of bacterial pathogens are known to cause serious infections of the CNS or PNS. Unfortunately, these infections are often serious and life threatening. Figure 8. summarizes some important infections of the nervous system.
Key Concepts and Summary
- Bacterial meningitis can be caused by several species of encapsulated bacteria, including Haemophilus influenzae, Neisseria meningitidis, Streptococcus pneumoniae, and Streptococcus agalactiae (group B streptococci). H. influenzae affects primarily young children and neonates, N. meningitidis is the only communicable pathogen and mostly affects children and young adults, S. pneumoniae affects mostly young children, and S. agalactiae affects newborns during or shortly after birth.
- Symptoms of bacterial meningitis include fever, neck stiffness, headache, confusion, convulsions, coma, and death.
- Diagnosis of bacterial meningitis is made through observations and culture of organisms in CSF. Bacterial meningitis is treated with antibiotics. H. influenzae and N. meningitidis have vaccines available.
- Clostridium species cause neurological diseases, including botulism and tetanus, by producing potent neurotoxins that interfere with neurotransmitter release. The PNS is typically affected. Treatment of Clostridium infection is effective only through early diagnosis with administration of antibiotics to control the infection and antitoxins to neutralize the endotoxin before they enter cells.
- Listeria monocytogenes is a foodborne pathogen that can infect the CNS, causing meningitis. The infection can be spread through the placenta to a fetus. Diagnosis is through culture of blood or CSF. Treatment is with antibiotics and there is no vaccine.
- Hansen’s disease (leprosy) is caused by the intracellular parasite Mycobacterium leprae. Infections cause demylenation of neurons, resulting in decreased sensation in peripheral appendages and body sites. Treatment is with multi-drug antibiotic therapy, and there is no universally recognized vaccine.
License
This work is produced by OpenStax-CNX and licensed under the Creative Commons Attribution License 3.0 http://cnx.org/content/m46535/1.3/
