2.4 Chronic Neurodegenerative Diseases
Alpha-Synucleinopathies and Parkinson’s Disease
The current model for most neurodegenerative conditions is that neurons die via apoptosis or a programmed form of cell death. This has very important clinical implications as turning on or turning off specific genes/proteins is a method to stop or possibly reverse neurodegeneration. Neurodegenerative diseases such as Parkinson’s Disease and Alzheimer’s Disease and others are all believed to use apoptotic pathways and additionally suggest that there is a component protein that is aberrantly folded to produce apoptosis.
Parkinson’s Disease: Overview
Most cases of this disorder appear to be sporadic (i.e. non-genetic in origin). A very few cases seem to have a genetic origin (for example the genes most often associated with DJ-1, Parkin (Ubiquitin E3 ligase) and alpha synuclein genes). Parkinson’s Disease is most often associated with loss of “pigmented” nuclei in the brain and typically involve the loss of a group of neurons found in the Substantia Nigra (Figure 1.). The substantia nigra neurons are dopaminergic and are pigmented because they contain the protein melanin.
The typical onset for Parkinson’s Disease is middle to later stages of life (i.e. 50 and beyond) although a small percentage of individuals who have known genetic mutations in Parkin or alpha-synuclein develop symptoms earlier.
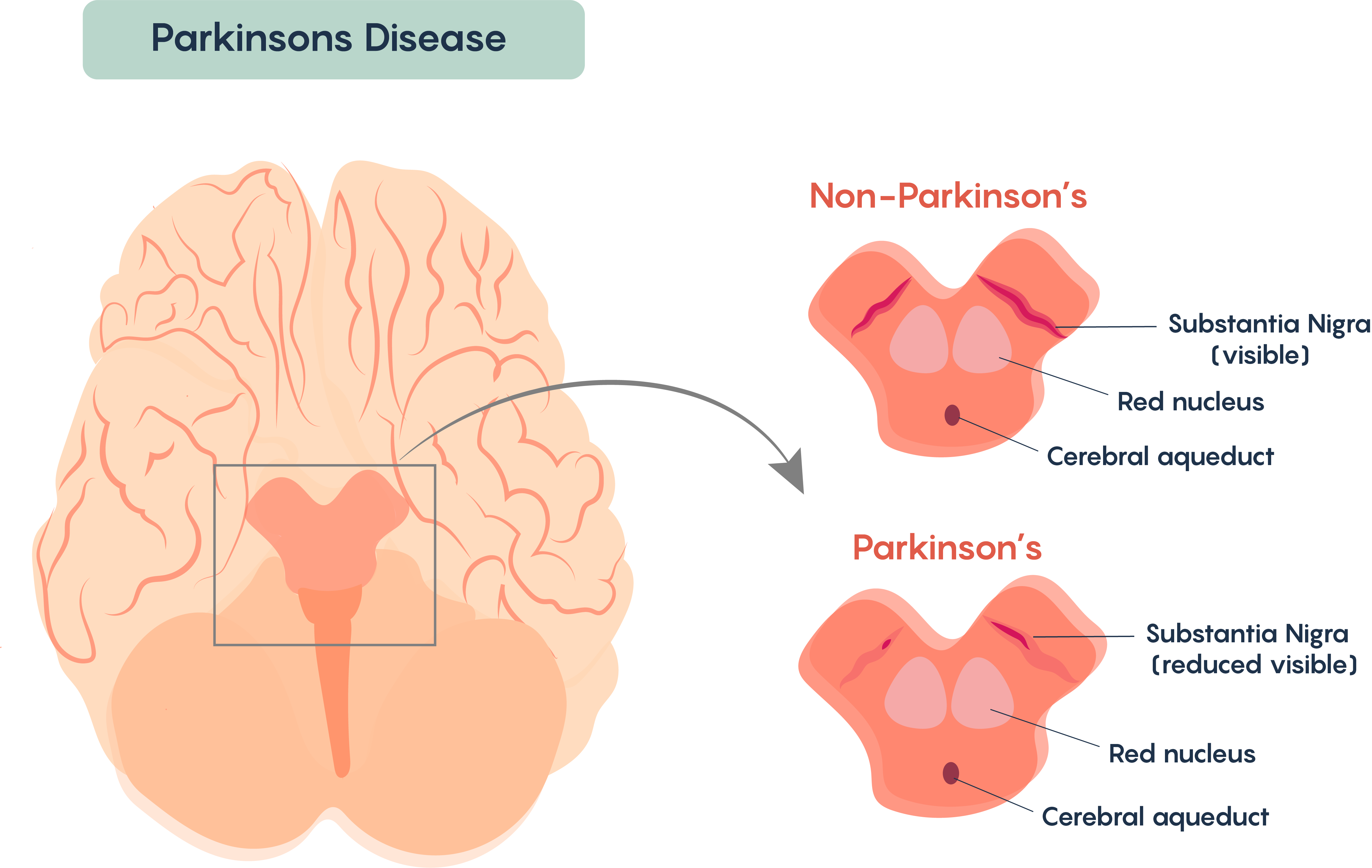
Loss of these cells affect the processing and execution of voluntary movement in individuals with Parkinson’s Disease. Similar to Multiple Sclerosis, once diagnosed, the symptoms become continuous and progressive – i.e. the symptoms worsen over time. Again, there are many similarities with Multiple Sclerosis, and there is no known cure for Parkinson’s Disease and the disease remains idiopathic although there are some known causes of Parkinson’s Disease including loss of movement following cerebral atherosclerosis, viral encephalitis, and as the result of side effects from drugs such as phenothiazides and reserpine.
“Classic” symptoms associated with Parkinson’s Disease
- Bradykinesia: Slowness in Initiation and Execution of Voluntary Movements
- Rigidity: Increase Muscle Tone and Increase Resistance to Movement (Arms and Legs Stiff) – as severity increases produce cogwheel rigidity
- Tremor: Usually Tremor at Rest; When person sits, arm shakes; Tremor Stops when person attempts to grab something (pill rolling tremor)
- Postural Instability: abnormal fixation of posture (stoop when standing), problems with equilibrium, and righting reflex
- Gait Disturbance: Shuffling feet
- Orthostatic Hypotension
- Dementia (in some instances)
- Dystonia (inappropriate and continuous muscle contraction)
- Ophthalmoplegia (weakness in eye muscles)
- Affective Mood Disorders (such as major depression)
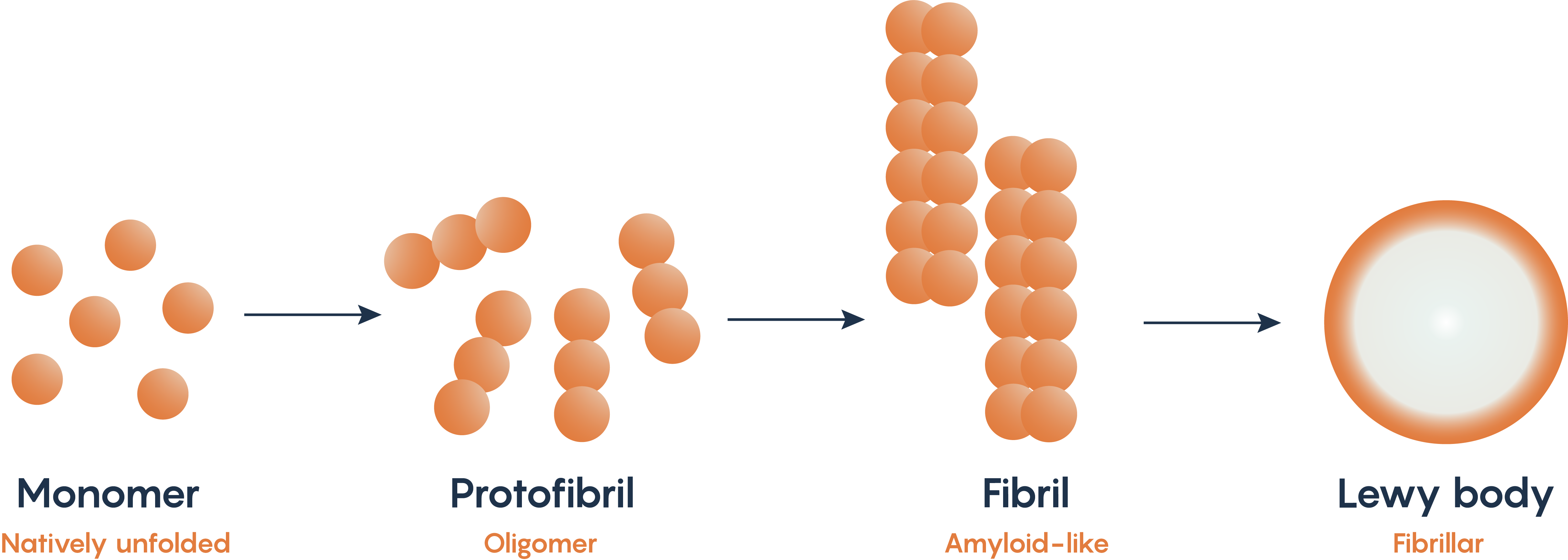
Alpha-synuclein is a naturally occurring protein within neurons. Mutations in the PARK1 and PARK4 genes which normally encodes for alpha-synuclein, have been associated with Parkinson’s Disease. As such, many animal models of Parkinson’s Disease look for the production of the fibrillary form of alpha-synuclein as it misfolds and then accumulates within the substantia nigral neurons known as Lewy bodies (Figure 2.). The misfolding and accumulation of alpha-synuclein has been hypothesized to be the reason that neurons undergo apoptosis, although the exact mechanism for how this occurs remains to be elucidated.
Animal Models of Parkinson’s Disease
The lack of candidate genes (except for alpha-synuclein, Parkin and DJ-1) has meant that most scientists have looked at toxin models. Most of the toxins that produce features that resemble changes in movement as well as Lewy-body like formation use catecholaminergic destruction. In fact, most of these toxins are incredibly powerful and dangerous mitochondrial complex inhibitors such as reserpine, MPTP, methamphetamine, 6-OH-dopamine, rotenone, and paraquat). In fact one toxin, MPTP is a by-product of synthetic heroin production suggesting that there may be a synthetic substance that causes Parkinson’s Disease and some epidemiological studies show a correlation with pesticide (such as paraquat and rotenone) usage and Parkinson’s Disease.
Alzheimer’s Disease: Overview and General Pathology
Another neurodegenerative condition is Alzheimer’s Disease (AD). This neurodegenerative condition is a form of dementia characterized the most common symptoms associated with Alzheimer’s Disease notably memory loss, problems with communication and difficulty finding words, attention problems, confusion and spatial disorientation, decreased or poor judgement, changes in mood and personality.
Affected areas explain the AD pathology, which is characterized by memory loss due to shrinkage of the hippocampus, and other problems which include higher level thinking and performance which are controlled by the cortex. Brain atrophy typically begins in the medial temporal lobe (i.e. hippocampus), moves on to the association cortices and therefore affects sensory and motor areas. It also affects the Nucleus Basalis of Meynert which has multiple cholinergic projections to the cortex (and is thought to be responsible for control of sleep, attention, and consciousness) as highlighted in Figure 3.
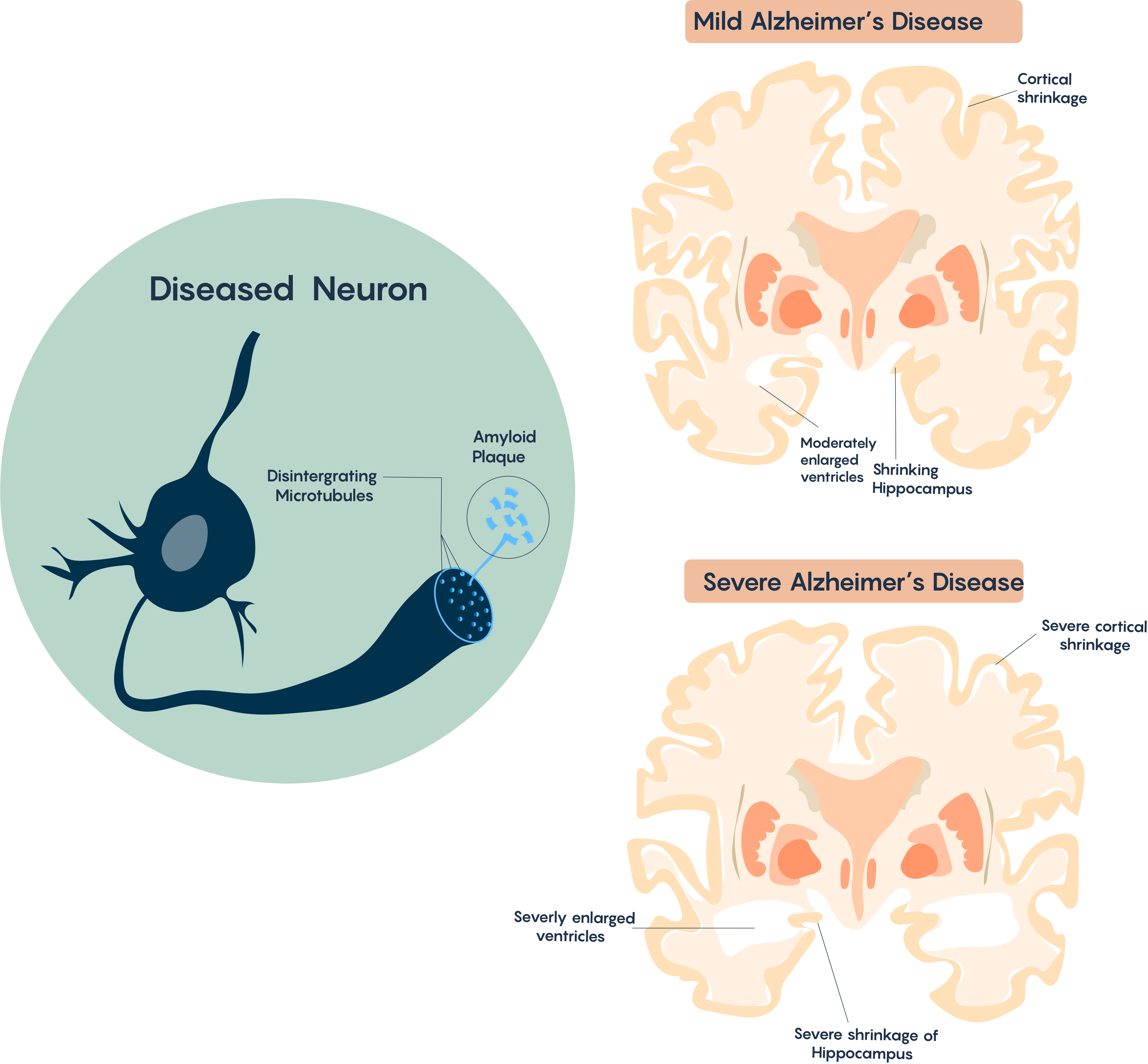
Cellular Pathology
The typical AD brain is characterized by the loss of neurons, progressive loss of synapses and cholinergic projections, the accumulation of extracellular β-amyloid (Aβ) plaques and intracellular neurofibrillary tangles (NFTs). With increased time and pathology this will often correlate with the formation of a glial scar (involving reactive astrocytes). There is also very strong evidence that this disorder may also have a strong immune component as there is often the infiltration of microglial cells in the Alzheimer’s Disease brain.
β-amyloid (Aβ) plaques: βA Cascade hypothesis
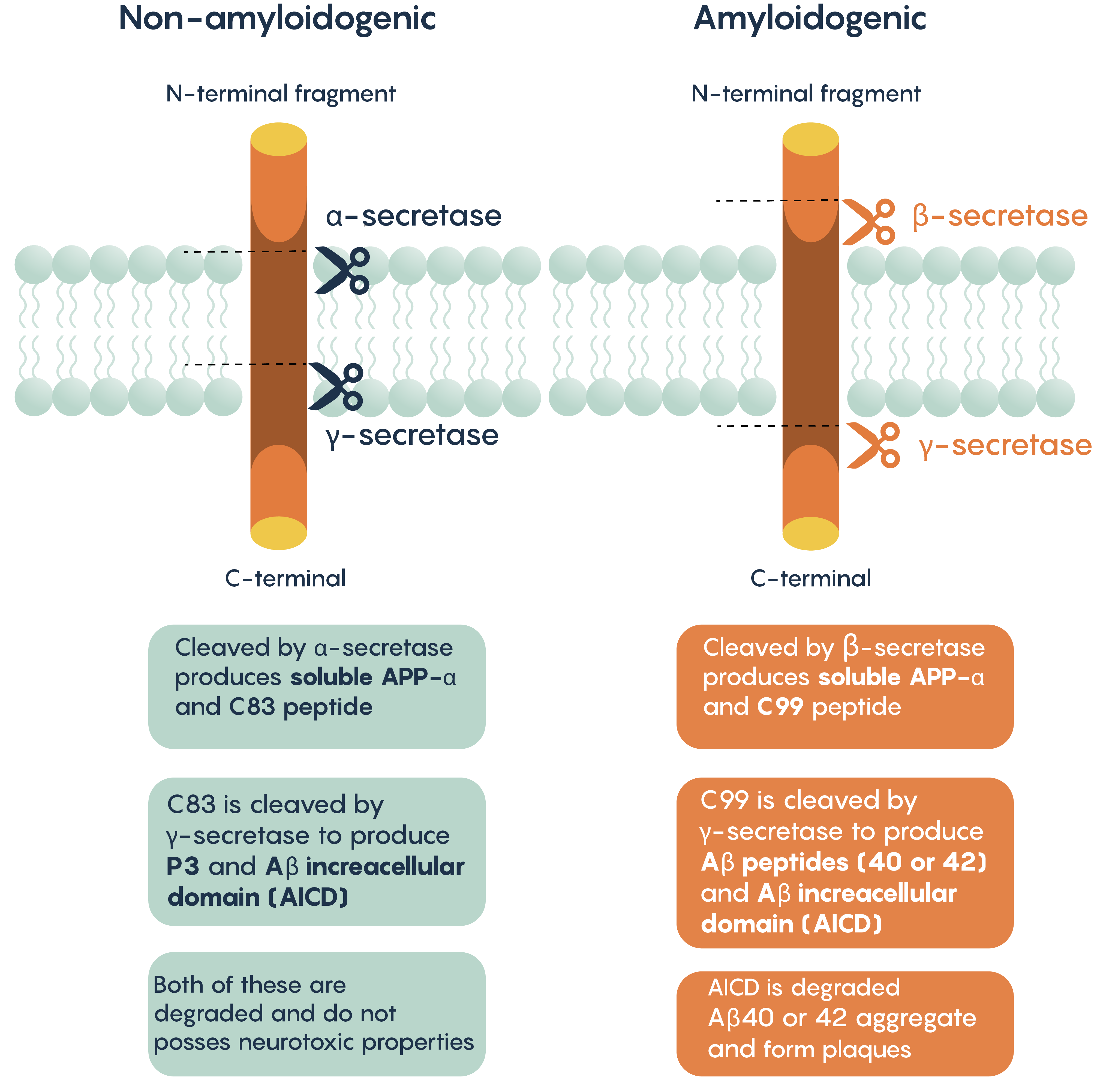
One of the hallmark histopathological and molecular features of Alzheimer’s Disease is the presence of extracellular aggregates that include -amyloid plaques. These plaques are believed to induce cytotoxicity by disrupting normal neuronal functions, ion concentrations, action potential generation. As with the modern theory of neurodegeneration, these plaques are composed of the cleaved form of amyloid precursor protein (APP).
APP can be processed via two different pathways:
- Non-amyloidogenic pathway (cleaved by α- and γ-secretases)
- Amyloidogenic pathway (cleaved by β- and γ-secretases)
Neurofibrillary Tangles (NFTs)
Another feature of the Alzheimer’s Diseased brain is the appearance of neurofibrillary tangles (NFTs) that are composed of hyper-phosphorylated tau protein. Normally within neurons (and in fact most cells), tau binds to and stabilizes microtubules (MTs), however, when this is phosphorylated by kinases, tau loses its affinity to MTs and dissociates from the MT complex. This causes the microtubules in turn to disassemble which interferes with proper axonal transport and eventually leads to loss of neuronal integrity. As such the Phospho-tau proteins form intracellular aggregates (NFTs) that become another defining feature of this disease as well as a potential target for therapeutic intervention.
Genetics of Alzheimer’s Disease
While sporadic AD cases account for the majority of AD patients, familial AD (FAD) accounts for only 5% of them. The familial form, FAD manifests earlier (around 40-50 years of age) compared to >65 years of age in sporadic cases. Despite this drastic difference in disease onset, symptoms are identical. Only 50% of FAD cases can be explained by known mutations in genes encoding APP and presenilins 1 and 2.
APP mutations are relatively rare and is characterized by individuals showing symptoms with an average age onset in their early 50’s. The conclusion that APP mutations might cause Alzheimer’s Disease is based on the observation that most Down Syndrome patients also develop AD after age 40 since they have an extra copy of Ch21 (which codes for APP). However, most inherited forms of APP mutations alter APP processing to:
- increase cleavage via the β-secretase pathway
- increase the Aβ42/40 ratio
- therefore, lead to production of peptides with higher fibrillogenic potential (Figure 2.)
