39 CASE 2- 2024: A 9-YEAR-OLD WITH RECURRENT RESPIRATORY INFECTIONS
A case report illustrating the importance of access to cystic fibrosis care and therapy. doi.org/10.4103/jopp.jopp_2_24. Ycaza-Zurita, M. G., Keating, C., & Sadeghi, H.
Clinical Summary 1
A 9-year-old Hispanic girl, previously diagnosed with asthma, was evaluated for frequent respiratory infections, chronic productive cough, poor growth, and gastrointestinal issues such as foul-smelling stools and post-tussive vomiting. Multiple hospitalizations for pneumonia were noted.
Physical examination revealed severe malnutrition (BMI below the 3rd percentile), digital clubbing, and diffuse lung crackles. Laboratory tests showed elevated white blood cell count, increased liver enzymes, deficiencies in fat-soluble vitamins, pancreatic insufficiency, and a positive sweat chloride test (103 mmol/L). Genetic analysis identified two copies of the F508del mutation in the CFTR gene. Imaging demonstrated hyperinflated lungs with peribronchial thickening and bronchiectasis, while pulmonary function tests revealed mixed obstructive-restrictive disease (FEV₁ 48%, FVC 60% predicted).
Initiation of a multidisciplinary care plan, including nutritional support and pulmonary therapies, resulted in clinical improvement and stabilization.
Learning Objectives
- Understand cystic fibrosis pathophysiology and its multi-organ involvement.
- Identify clinical features and diagnostic markers of cystic fibrosis.
- Interpret laboratory, imaging, and genetic testing results to support the diagnosis of cystic fibrosis.
Clinical History1
- Age: 9 years old
- Sex: Female
- Ethnicity: Hispanic
Medical History1
- Initially diagnosed with asthma at age 2
- Multiple hospitalizations for pneumonia
- No family history of cystic fibrosis (CF)
- Poor growth despite adequate nutrition
Symptoms1
- Recurrent respiratory infections
- Persistent productive cough and chest congestion
- Foul smelling bowel movements and post-tussive vomiting
- Daytime lethargy and failure to gain weight
Physical examination1
- Severe malnutrition with body mass index (BMI <3rd percentile)
- Digital clubbing
- Diffuse lung crackles
- Vital signs stable; oxygen saturation 97% on room air
Laboratory investigations1
| Test | Result | Reference Range | Interpretation |
| WBC | 15.23 × 10³/μL | 4–11 × 10³/μL | Elevated, indicative of infection or inflammation |
| AST | 78 U/L | 10–40 U/L | Elevated, CF-related liver disease |
| ALT | 181 U/L | 7–56 U/L | Elevated |
| ALP | 405 U/L | 44–147 U/L | Elevated |
| GGT | 125 U/L | 8–61 U/L | Elevated |
| Vitamin D | 14 ng/mL | 30–100 ng/mL | Deficient |
| Vitamin E | 2.3 mg/L | 5.5–9.0 mg/L | Deficient |
| Pancreatic fecal elastase | <10 μg/g | >200 μg/g | Severe pancreatic insufficiency |
| Sweat chloride | 103 mmol/L | <60 mmol/L | Positive for CF |
WBC = White Blood Cells; AST = Aspartate Aminotransferase; ALT = Alanine Aminotransferase; ALP = Alkaline Phosphatase; GGT = Gamma-Glutamyl Transferase.
Microbiology1
- Sputum cultures were positive for non-mucoid Pseudomonas aeruginosa and methicillin-sensitive Staphylococcus aureus (MSSA).
- Later, Mycobacterium avium complex (MAC) was detected, but she was clinically stable.
Imaging1
Chest X-ray:
Revealed hyperinflated lungs, diffuse peribronchial thickening, and predominantly upper-lobe bronchiectasis
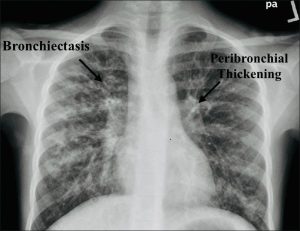
Figure 1: Anteroposterior chest X-ray: Classic changes of cystic fibrosis, with hyperinflated lungs, diffuse peribronchial thickening, and predominantly upper lobe bronchiectasis. https://doi.org/10.4103/jopp.jopp_2_24
Abdominal ultrasound
Showed mild liver echogenicity and intrahepatic bile duct dilation.
Pulmonary Function Tests (PFTs)1
Demonstrated a reduced forced expiratory volume in one second (FEV₁) of 0.86 L (48% predicted) and forced vital capacity (FVC) of 1.19 L (60% predicted), with a preserved FEV₁/FVC ratio of 72% and no bronchodilator reversibility. These findings indicate a mixed obstructive and restrictive ventilatory pattern consistent with CF-related lung disease.
Genetic Testing1
Targeted gene sequencing confirmed two F508del mutations, establishing a Cystic Fibrosis diagnosis.
Questions and Answers Leading to Diagnosis:
Question 1: Based on the presenting symptoms, what initial diagnoses should be considered?
Question 2: What is the underlying genetic cause of cystic fibrosis?
Question 3: How does Cystic Fibrosis affect pulmonary and gastrointestinal physiology (Pathophysiology in this patient)?
Question 4: Which laboratory investigations confirm the diagnosis, and what findings are diagnostic?
Question 4: What is the final diagnosis in this case?
** For answers please check the next chapter.**
Medical terminologies.
- Cystic Fibrosis (CF) – A hereditary, life-limiting disorder caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene, affecting the lungs, pancreas, liver, and other organs1.
- CFTR Gene – The Cystic Fibrosis Transmembrane Conductance Regulator gene, located on chromosome 7q31.2, encodes a protein that functions as a chloride channel in epithelial cells.
- F508del Mutation – The most common CF-causing mutation involves the deletion of phenylalanine at position 508 of the CFTR protein, leading to misfolding and loss of function3.
- Autosomal Recessive Inheritance – A genetic pattern where two copies of a mutated gene (one from each parent) are required for the disease to manifest2.
- Sweat Chloride Test – A diagnostic test that measures the amount of chloride in sweat; values above 60 mmol/L are diagnostic of CF1,2.
- Pancreatic Insufficiency – Inability of the pancreas to secrete digestive enzymes, leading to poor nutrient absorption, malnutrition, and fatty stools (steatorrhea)1,2.
- Steatorrhea – Passage of bulky, oily, foul-smelling stools due to fat malabsorption2.
- Bronchiectasis- Permanent dilation and scarring of the bronchi are caused by chronic infection and inflammation2.
- Pulmonary Function Tests (PFTs) – A group of tests that assess lung function; in CF, these often show a mixed obstructive and restrictive pattern1,2.
- FEV₁ (Forced Expiratory Volume in 1 second)- The amount of air a person can forcefully exhale in one second; reduced in obstructive airway diseases2.
- FVC (Forced Vital Capacity) – The total amount of air exhaled during the FEV test; used alongside FEV₁ to evaluate lung function2.
References
- Ycaza-Zurita, M. G., Keating, C., & Sadeghi, H. (2024). Cystic fibrosis – A case report illustrating the importance of access to cystic fibrosis care and therapy. Journal of Pediatric Pulmonology, 3(1), 23–25. https://doi.org/10.4103/jopp.jopp_2_24
- Sankari, A., & Sharma, S. (2025). Cystic Fibrosis. In StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK493206/
- Ostedgaard, L. S., Meyerholz, D. K., Chen, J. H., Pezzulo, A. A., Karp, P. H., Rokhlina, T., … Stoltz, D. A. (2011). The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Science Translational Medicine, 3(74), 74ra24. https://doi.org/10.1126/scitranslmed.3001868
Bronchiectasis- Permanent dilation and scarring of the bronchi are caused by chronic infection and inflammation.
FEV₁ (Forced Expiratory Volume in 1 second)- The amount of air a person can forcefully exhale in one second; reduced in obstructive airway diseases.
FVC (Forced Vital Capacity) - The total amount of air exhaled during the FEV test; used alongside FEV₁ to evaluate lung function
