Sam’s Health Part A: Cystic Fibrosis
When Sam was 2 years and 6 months, Nancy & Paul were concerned about his newly developed symptoms:
- They could hear Sam wheeze when he was breathing
- Greasy/oily stools in his diaper
- Family physician referred Sam to a pediatrician

Initial Visit
Sam’s family physician orders the following tests and looks at his immunization record:
- Physical examination
- Blood work
- Chest x-ray (CXR)
Sam is referred to a pediatrician
Sam’s physician takes a look at his immunization
Sam’s immunization record was incomplete, serological testing was done to determine immunity.
Serology is only recommended in this situation for:
- Varicella
- Measles
- Mumps
- Rubella
- Hepatitis B
Physical Examination
Vital Signs
- Age: 2 years, 6 months
- Weight: 28 lbs. (lower 10%-tile)
- Height: 3 ft. 1 inch
- Pulse: 115 BPM
- Respirations: 30 breaths per minute
- Blood pressure: 95/60 mmHg
Abdominal
- No swelling present
Extremities
- Full mobility is present
- Pulse found in arms and legs
Genitourinary
- Not assessed
Neurological
- Normal reflexes
General Appearance
- Happy, energetic child
Head and Neck
- Runny nose but his ears are clear of fluid
- No enlarged lymph nodes in neck
Lungs
- Crackling sounds are present
- Coughing and wheezing are audible
Cardiovascular
- Normal
Sam’s blood work revealed
- White blood cell count: values within normal limits
- WBC differential: values within normal limits
- Red blood cell count: values within normal limits
- Hematocrit: values within normal limits
- Platelet count: values within normal limits
- Sweat chloride test: 40 mmol/L
- Chest x-ray: some hyperinflation & bronchial wall thickening
Normal CXR
- Bones are white in colour, calcification will appear white
- Normally lungs are full of air (black in colour)
- Fluid or blood are white in colour

Diagnosing Sam

- Nancy forgot to mention the colour of Sam’s sputum
- Sam is coughing up green, viscid sputum
- Sam had white ‘frosting’ on his face this visit
- Nancy stated: “that must be why his skin tastes a bit salty when I kiss him”
- The pediatrician ordered a sweat test be conducted and a CT scan done to confirm his suspicions
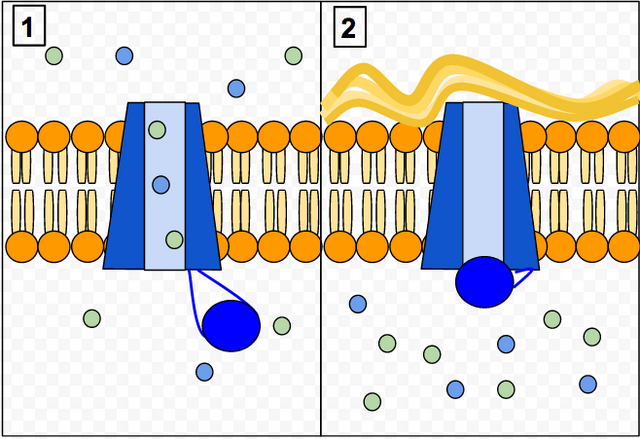
Understanding Sweat Test Results
Individuals with CF have more chloride in their sweat and will require a Sweat Chloride test to confirm the diagnosis
To understand what the sweat test results mean, a chloride level of:
- Less than or equal to 29 mmol/L = Unlikely
- Between 30-59 mmol/L = Possible
- Greater than or equal to 60 mmol/L = Likely
Sweat chloride test results fall between the range of 30-59 mmol/L, test is usually repeated.
Sam’s chloride levels: 65 mmol/L
Cystic Fibrosis
What is cystic fibrosis?
- Life-threatening genetic disease
- Body creates thick mucus and obstructs ducts U tubes in the lungs, digestive tract and pancreas
- Affects the sweat glands & male reproductive system
Diagnosis for Cystic Fibrosis
- Newborn screening: Genetic test to screen newborns for the disease
- Chest X-ray: Study the effect of disease on the heart & lungs
- Prenatal screening: Amniocentesis test to check for CF in fetus
- Sputum culture: To detect presence of bacteria pseudomonas
- Cystic fibrosis carrier testing: Allows parents to find out what their chances are of having a child with CF
Newborn Screening
Newborn screening (NBS) for cystic fibrosis is done in the first few days after birth. By diagnosing CF early, CF health care providers can help parents learn ways to keep their child as healthy as possible and delay or prevent serious, lifelong health problems related to CF.
Research shows that children who receive CF care early in life have better nutrition and are healthier than those who are diagnosed later. Early diagnosis and treatment can:
- Help keep lungs healthy
- Reduce hospital stays
- Add years to life
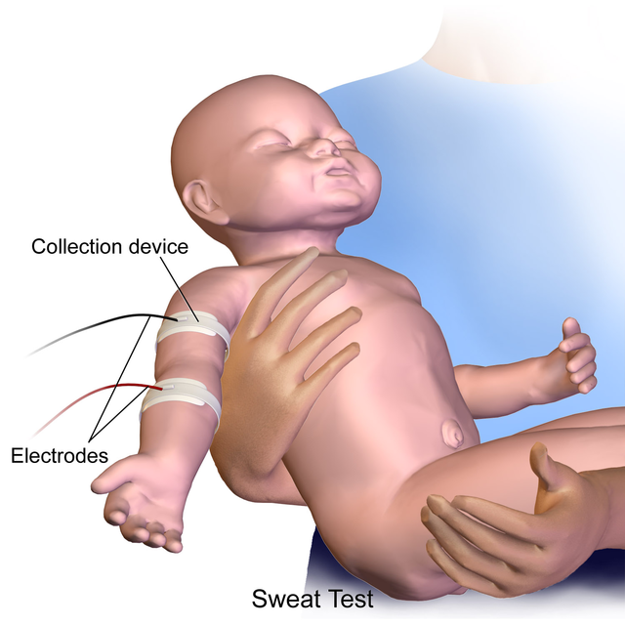
How is Newborn Screening Done?
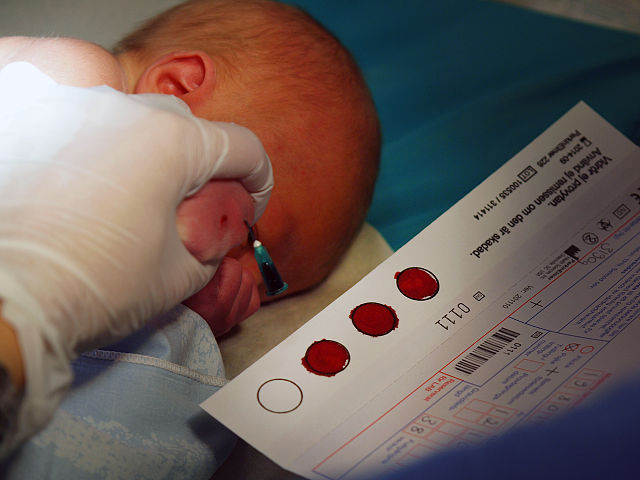
Newborn screening is done during the first few days of a baby’s life.
A few drops of blood from a heel prick are placed on a special card, called a Guthrie card.
This card is then mailed to a special state laboratory that will test the blood sample for certain health conditions, including CF. In some states, newborn screening involves two blood samples, one at birth and one a few weeks later.
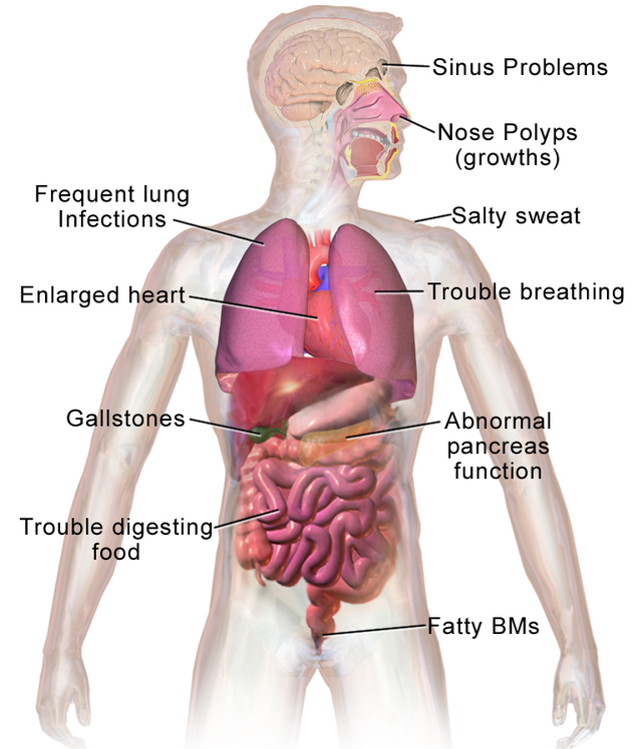
Symptoms of Cystic Fibrosis
CC-BY-3.0
Sam’s Complications
- After hearing about Sam’s diagnosis Nancy and Paul do their own research and try to learn as much as they can about Sam’s quality of life.
- They learn that CF can lead to many complications.
Nancy and Paul do research on Sam’s diagnosis
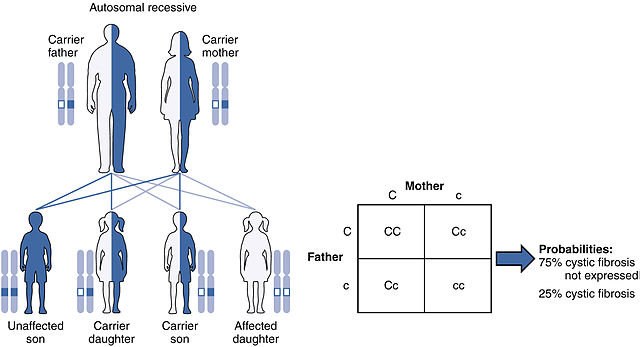
Autosomal recessive
- 1 copy of mutated gene inherited 🡪 CF carrier
- 2 copies of mutated gene inherited 🡪 CF
- Carriers can pass their copy of the mutated gene to their children
Complications of Cystic Fibrosis
Respiratory System
- Recurrent attack of bronchitis & pneumonia
- Damage to the airways → bronchiectasis
- Respiratory failure
Digestive System
- Malabsorption of nutrients
- Diabetes mellitus (pancreatic damage)
- Blockage of bile duct
Reproductive System
- Infertility
- Pregnancy may exacerbate the disease & cause complications
Other Systems
- Increased risk of osteoporosis
- Dehydration & electrolyte imbalances
- Clubbing of fingers & toes
Complications of Cystic Fibrosis – Histological Findings
| Tissue | Stain | Pathological findings | H&E: Hematoxylin and eosin stain |
| Lung | H&E |
|
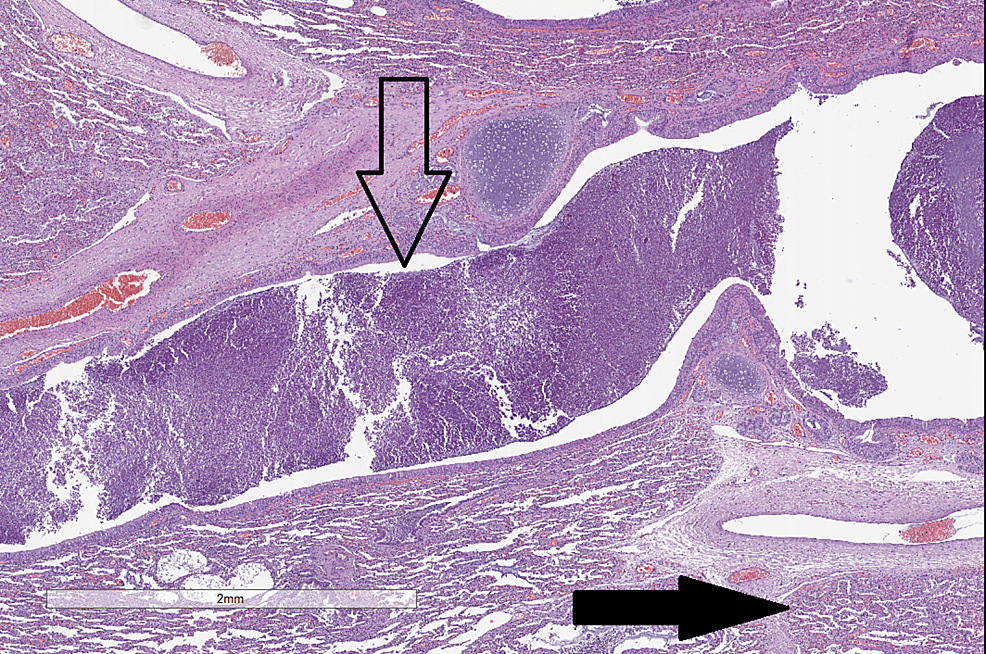
CC-BY 3.0 |
| Liver | H&E |
|
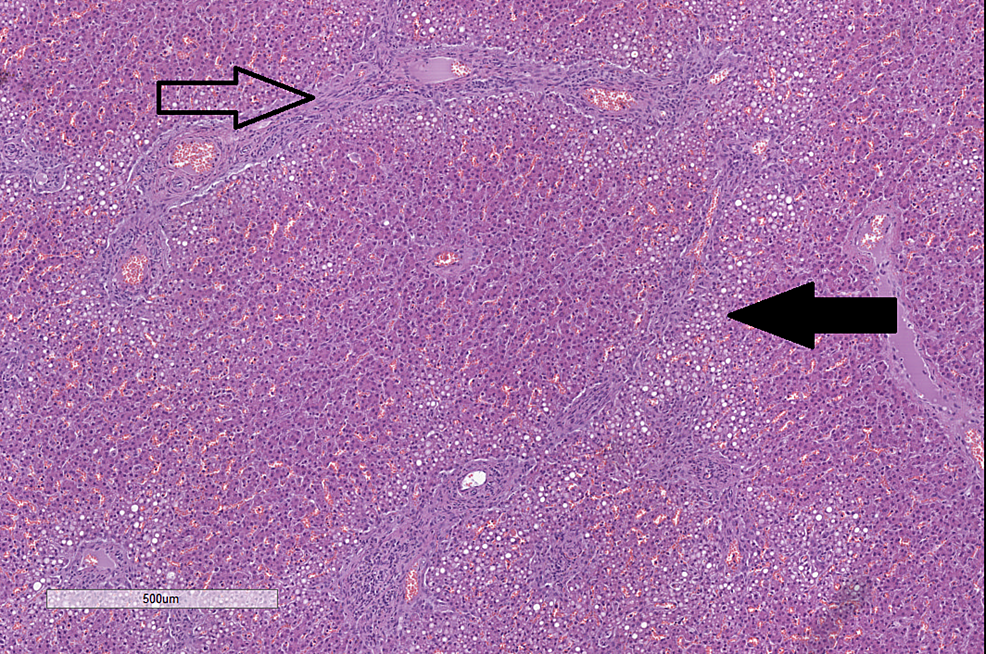
CC-BY 3.0 |
| Pancreas | H&E |
|
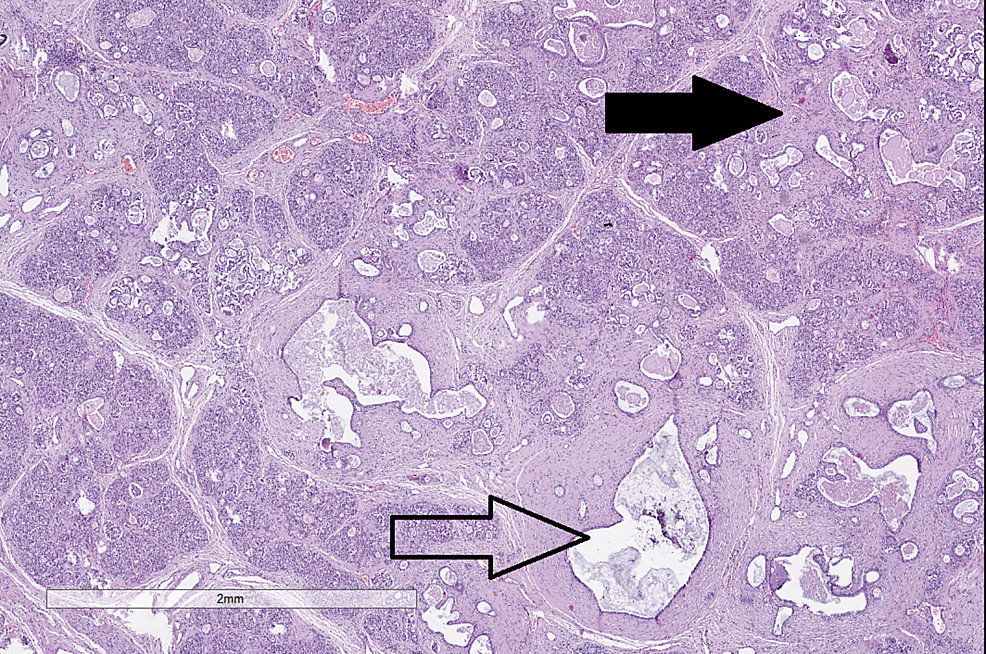
CC-BY 3.0 |
| Small bowel | H&E | Thick mucous secretions in glands | 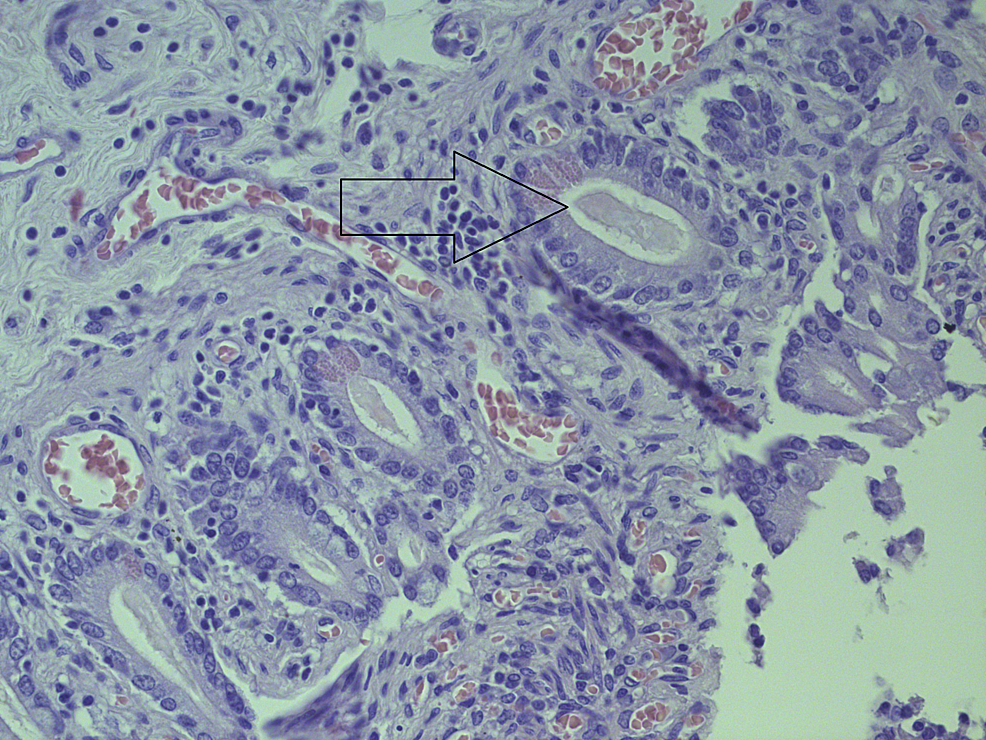
CC-BY 3.0 |
| Kidney | H&E |
|
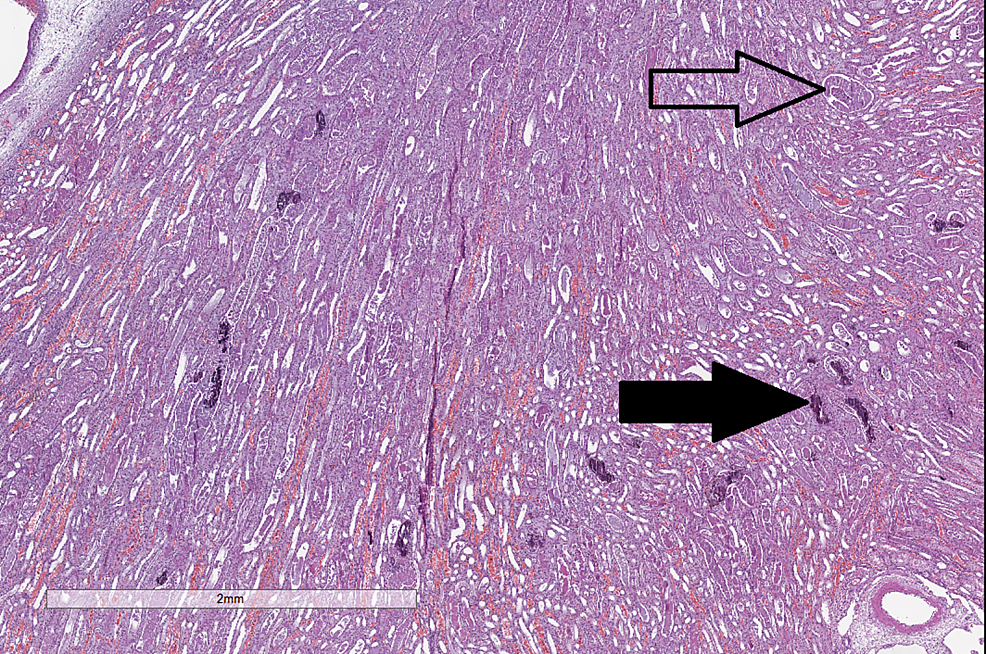
CC-BY 3.0 |
Treating Sam’s CF
Sam’s pediatrician explains to Nancy and Paul that at present there is no cure for CF.
Sam’s pediatrician recommends treatment aimed at alleviating the symptoms & reducing the incidence of complications.
How do you Treat Cystic Fibrosis?
Careful follow-up and early & aggressive intervention is essential. It is recommended to obtain treatment at a centre that specializes in CF.
Aims of Treatment
- Reducing incidence & successful treatment of respiratory infections
- Providing sufficient nutrition
- Removal of & loosening mucus plugs in lung & airways
Chest Physical Therapy
- Loosens thick mucus & aids in expectoration
- Aerobic exercise helps loosen the mucus
- Breathing exercises – coughing or huffing
Q&A
How does physical activity support children with CF to manage their disorder?
Physical activity and sports will assist CF clients in preserving their lung capacity long term and decreases the incidence of complications related to CF such as frequent lung infections, respiratory failure, delayed growth and weight loss, digestive disorders and malabsorption of the bowel. Participation in sports often encourages children and adolescents to perform and achieve goals in advancing in their respective activities, this often requires a regime to practice daily and have a healthy diet (these habits reduce the risk of infection)
Nutritional Therapy
Nutritional therapy can address malnutrition & vitamin deficiency that is common to CF patients. It involves:
- Intake of oral pancreatic enzymes to help in digestion & absorption
- Vitamin A, D, E & K supplements
- High-calorie snacks/meals
Q&A
What type of diet may a child with Cystic Fibrosis need?
There are little dietary restrictions in clients living with CF and they often requires a high protein, high fat, high calories, and vitamin/ mineral rich diet to compensate for their nutritional deficiencies. Refer to Canada’s Food Guide (2019) for acceptable eating habits for children
Medication
Antibiotics
Treating & preventing respiratory infections
Mucolytics
- Aid in coughing out mucus
- Improves lung function
Bronchodilators
- Keep airways patent
- Facilitate easier breathing

CF complications and surgical intervention
- Removal of mucus obstructing airways (endoscope)
- Relieving intestinal obstruction
- Lung transplant