N05: Les données à haute-dimension
Maheshwar Panday
Données sur le cancer du sein au Wisconsin : Analyse des données à haute dimension
Vous êtes pathologiste et vous avez mesuré 569 noyaux de cellules à partir de prélèvements à l’aiguille de masses de tissu mammaire. Les échantillons proviennent de masses bénignes (B) ou malignes (M). Vous souhaitez effectuer une analyse de la forme et de la taille des cellules entre les cellules bénignes et malignes afin de mieux comprendre les différences qui existent entre elles. Vous aimeriez également explorer l’utilisation de l’apprentissage automatique pour voir dans quelle mesure de simples algorithmes de regroupement peuvent identifier les cellules bénignes des cellules malignes en se basant UNIQUEMENT sur leurs caractéristiques de taille et de forme. Utilisation de l’ensemble de données sur le cancer du sein du Wisconsin
—- Texte tiré de Kaggle ——-
Les données sur le cancer du sein comprennent 569 exemples de biopsies cancéreuses, chacune comportant 32 caractéristiques. Une caractéristique est un numéro d’identification, une autre est le diagnostic du cancer et 30 sont des mesures de laboratoire à valeur numérique. Le diagnostic est codé “M” pour malin ou “B” pour bénin.
Les 30 autres mesures numériques comprennent la moyenne, l’erreur standard et la valeur la plus mauvaise (c’est-à-dire la plus grande) pour 10 caractéristiques différentes des noyaux cellulaires numérisés, qui sont les suivantes:-
Rayon, Texture ,Périmètre, Surface, Lisse Compacité, Concavité, Concave Points, Symétrie ,Dimension fractale. Les données sont disponisbles du : https://archive.ics.uci.edu/dataset/17/breast+cancer+wisconsin+diagnostic
Pour en savoir plus sur l’ensemble de données du Wisconsin sur le cancer du sein, et plus particulièrement sur la manière dont chacune des caractéristiques de cet ensemble de données a été calculée, veuillez consulter :
Street, W.N., Wolberg, W.H., & Mangasarian, O.L. (1993). Nuclear feature extraction for breast tumor diagnosis. Electronic imaging.
(1) chargement de progiciels de statistiques, de regroupement, de réduction de la dimensionnalité et de visualisation des données
Installez et chargez les progiciels suivants dans votre environnement de RStudio.
library(dabestr) # estimation statistics
library(ggplot2) # plotting and data visualisation
library(pheatmap) # generating a heatmap
library(tidyverse) # data handling
library(dplyr) # data handling
library(stats) # basic statistics
library(RColorBrewer) # colour palettes and plot aesthetic controls
library(Rtsne) # for performing T-distributed stochastic neighbour embedding (tsne)
(2) Chargement des données
Téléchargez les données à partir du UCI Machine Learning Repository. Charger ensuite le fichier csv des mesures des cellules dans votre environnement de RStudio.
BreastCancer.Data <- read.csv("WisconsinBreastCancerData.csv")
BreastCancer.Data$X <- NULL
print (head(BreastCancer.Data))(3) préparer les cadres de données pour le tsne et le regroupement ultérieur
L’ensemble de la base de données BreastCancer.Data est structuré de telle sorte que chaque ligne correspond à une cellule et chaque colonne à un paramètre. Toutefois, certaines colonnes ne sont pas des caractéristiques (descriptions quantitatives des cellules). Lorsqu’une colonne n’est pas une caractéristique, il s’agit d’une étiquette. Les étiquettes sont des moyens d’identifier ou de marquer des cellules spécifiques une fois que nous avons compris comment elles sont caractérisées par leurs caractéristiques.
Avant de pouvoir explorer l’ensemble de données, nous devons séparer les étiquettes des caractéristiques. Deux colonnes du cadre de données étiquettent les cellules – id -> le numéro d’identification unique attribué à une cellule – diagnostic -> si la cellule provient d’un échantillon bénin (B) ou malin (M)).
Les colonnes restantes sont des caractéristiques qui servent de descriptions quantitatives de la taille et de la forme des cellules.
- Votre première tâche avec ces données est de subdiviser l’ensemble de la base de données sur le cancer du sein en deux bases de données
- Diagnostic.Labels – qui contiendra les métadonnées
- Diagnostic.Features – qui contiendra les caractéristiques
- Étant donné que les amplitudes des mesures s’étendent sur des échelles différentes, vous devez placer vos données dans un espace commun pour permettre aux variations et aux différences de devenir apparentes entre les caractéristiques de l’ensemble de l’ensemble des données. Obtenez la cote-Z votre ensemble de données à l’aide de l’opération scale() dans R sur votre Diagnostic.Features.
## ---------------------------------------- ##
##### Setting features apart from labels #####
## ---------------------------------------- ##
# labels dataframe - contains the unique identifier and the diagnosis)
Diagnostic.Labels <- select(BreastCancer.Data, id, diagnosis)
# features dataframe - all the columns that are not identifiers are the measured features that characterise the tissue mass - specifically the characterizing the nuclei present.
feature.columns <- setdiff (colnames(BreastCancer.Data), colnames(Diagnostic.Labels))
Diagnostic.Features <- BreastCancer.Data[, feature.columns]
# create a z-scored version of the features
Diagnostic.Features.Scored <- scale(Diagnostic.Features)
# print (head(Diagnostic.Features.Scored))
view(Diagnostic.Features.Scored)
print (colnames(Diagnostic.Features.Scored)(4) explorer l’ensemble de données à l’aide de t-SNE
Tsne ou le T-distributed Stochastic Neighbour Embedding est une technique utilisée pour visualiser des données de haute dimension en 2 dimensions. Qualifiée de méthode de réduction de la dimensionnalité, elle nous permet d’explorer les données une caractéristique à la fois. Dans cet ensemble de données sur le cancer du sein, 32 mesures décrivent la taille, la forme et la texture des masses dans le tissu mammaire, qui sont ensuite considérées comme bénignes (B) ou malignes (M).
Pour plus d’informations sur T-sne, veuillez consulter : Van der Maaten, L., Hinton, G. Visualizing Data using T-sne. Journal of Machine Learning Research 9 (2008) 2579-2605
Utilisons tsne pour visualiser la répartition de ces caractéristiques dans l’ensemble des données.
N.B. J’ai exécuté cette boucle for de combinaisons de semences par perplexité car il est important de tester ces paramètres lorsque l’on travaille avec T-sne. La perplexité contrôle la part des éléments de la structure de données locale par rapport aux éléments de la structure de données globale qui contribuent à la visualisation finale lorsque les données passent par la réduction de la dimensionnalité. Les variations de perplexité pour la même graine aléatoire peuvent avoir des effets très importants sur la visualisation finale, même si les données ne changent pas.
# Define your perplexities and seeds
perplexities <- c(10, 15, 20, 25, 30)
seeds <- c(123, 456, 789, 246, 135 )
# Create an empty dataframe to store the t-SNE components
tsne_data <- data.frame()
# Iterate over each combination of perplexity and seed
for (i in 1:length(perplexities)) {
for (j in 1:length(seeds)) {
# Perform t-SNE with the current perplexity and seed
tsne_model <- Rtsne(Diagnostic.Features.Scored, perplexity = perplexities[i], seed = seeds[j])
# Create a dataframe for the t-SNE components
tsne_temp <- data.frame(
tSNE1 = tsne_model$Y[, 1],
tSNE2 = tsne_model$Y[, 2],
Perplexity = perplexities[i],
Seed = seeds[j],
Diagnosis = Diagnostic.Labels$diagnosis
)
# Append the data to the main dataframe
tsne_data <- rbind(tsne_data, tsne_temp)
}
}
# Create the plot
tsne.stampcollection <- ggplot(tsne_data, aes(x = tSNE1, y = tSNE2, colour = Diagnosis)) +
geom_point() +
facet_wrap(Perplexity ~ Seed, scales = "free") +
theme_bw()
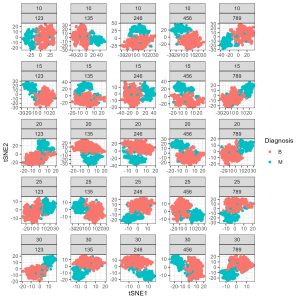
(4) Exercice de l’étudiant – tsne pour explorer les cellules bénignes vs malignes en utilisant la réduction de dimensionnalité
Utilisez les paramètres suivants dans votre tsne initial : – seed <- 789 – perplexité <- 30
Construisez une carte T-sne de la base de données Diagnostic.Features. Tracez la carte T-sne résultante à l’aide de ggplot en veillant à colorer les points en fonction de leur étiquette de diagnostic. Que remarquez-vous sur la carte T-sne lorsqu’elle est annotée par diagnostic ?
En guise d’exercice, variez la perplexité pour la graine de 789, en essayant d’aller de 10 à 50 par sauts de 10. Que remarquez-vous sur la carte T-sne lorsque vous faites varier la perplexité ? Selon vous, que contrôle le paramètre de perplexité dans l’algorithme T-sne ?
# Set your perplexity and seed
set.seed (789)
perplexity <- 30
seed <- 789
# Perform t-SNE with the specified perplexity and seed
tsne_model <- Rtsne(Diagnostic.Features.Scored, perplexity = perplexity, seed = seed)
# Create a dataframe for the t-SNE components
tsne_data <- data.frame(
tSNE1 = tsne_model$Y[, 1],
tSNE2 = tsne_model$Y[, 2],
Diagnosis = Diagnostic.Labels$diagnosis # Add the diagnosis column
)
# Load ggplot2
library(ggplot2)
# Create the plot
tsne.by.diagnosis <- ggplot(tsne_data, aes(x = tSNE1, y = tSNE2, colour = Diagnosis)) +
geom_point() +
labs( x = "T-SNE Component 1", y= "T-SNE Component 2")
theme_bw()
# Print the plot
print(tsne.by.diagnosis)
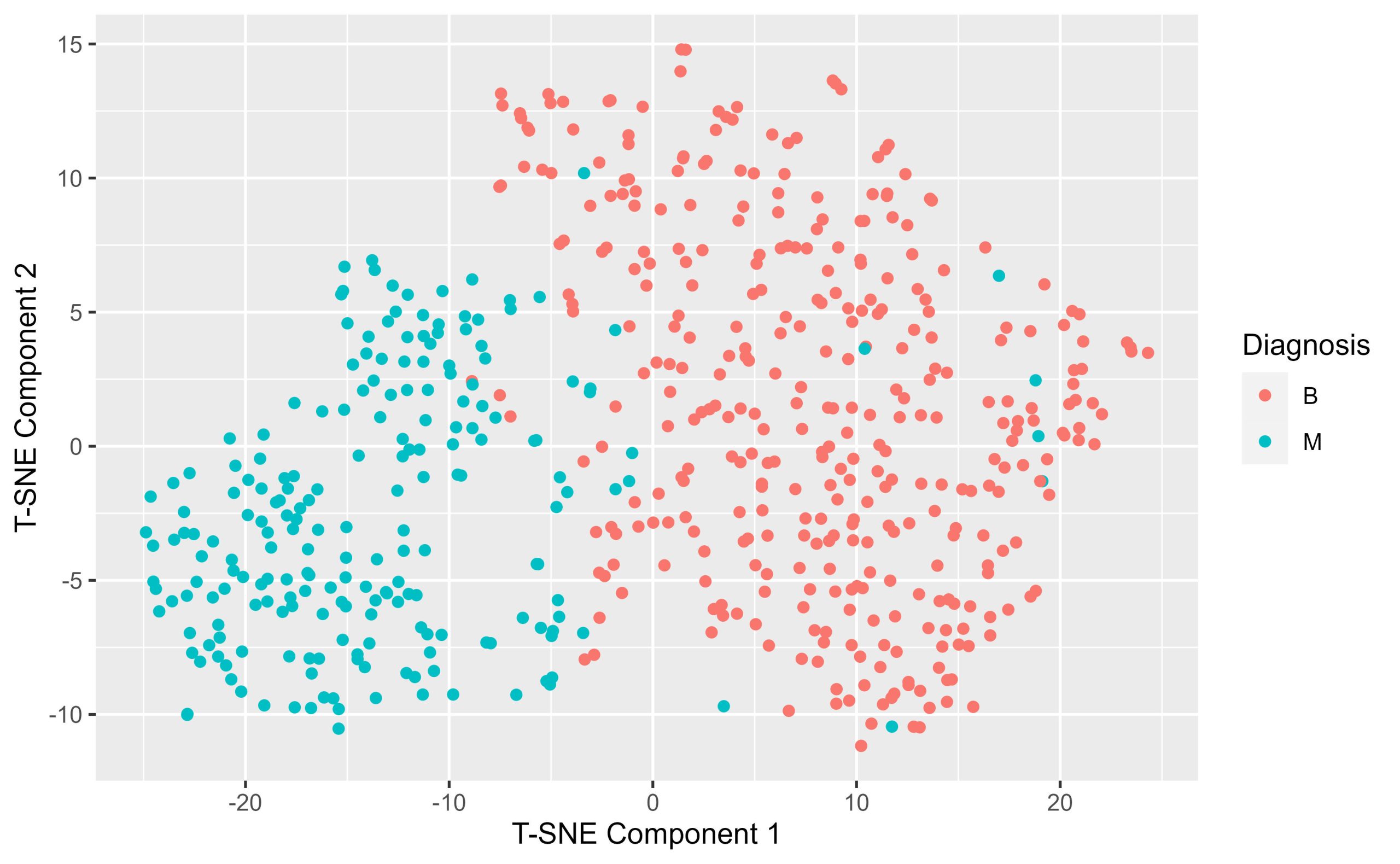
5) Regroupement hiérarchique d’échantillons malins
Et s’il existait un moyen d’identifier facilement les cellules comme étant bénignes ou malignes en fonction de leurs paramètres de taille et de forme ? Les algorithmes de regroupement sont un exemple d’approche fondée sur les données pour regrouper vos données sur la base des modèles présents dans les caractéristiques. Bien sûr, les données contiennent des étiquettes, mais que se passerait-il si vous faisiez des regroupements sur les valeurs des caractéristiques, les mesures, sans fournir d’informations sur le diagnostic des cellules ? Sélectionnez 2 grappes – nous voulons voir si le regroupement hiérarchique permet de distinguer les cellules bénignes des cellules malignes en se basant uniquement sur les informations relatives à leur forme et à leur taille.
Les étapes du regroupement hiérarchique sont les suivantes :
- Construire une matrice de dissimilarité des caractéristiques a partir des cotes- z. Utilisez les distances euclidiennes
- Appelez la fonction hclust sur la matrice de dissimilarité et spécifiez que la méthode est ward.D2
- Annotez le dendrogramme pour montrer où les données sont réparties en groupes (clusters)
- Visualisez les résultats de votre regroupement en construisant une carte thermique de la matrice de dissimilarité organisée avec un dendrogramme entouré et annoté.
## ----------------------------------------------------- ##
#### dissimilarity matrix and hierarchical clustering #####
## ----------------------------------------------------- ##
# set the number of clusters
num.clusters <- 2
# construct a pairwise dissimilarity matrix using euclidean distances
dissimilarity.matrix <- dist(Diagnostic.Features.Scored, method = "euclidean")
# hierarchical clustering with Wthe ward.D2 algorithm
h.clusters <- hclust(dissimilarity.matrix, method = "ward.D2")
# cut the dendrogram into k clusters
clusters <- cutree(h.clusters, k = num.clusters)
## --------------------------------------------------- ##
##### Preparing Dataframe for Downstream Statistics #####
## --------------------------------------------------- ##
# create a dataframe of identifiers and z-scored features
All.Samples.zscored <- cbind(Diagnostic.Labels,
Diagnostic.Features.Scored)
# append the clusters to the dataframe
All.Samples.zscored$hclust_clusters <- clusters
# view(Malignant.Samples.zscored) # inspection step
##--------------------------------------- ##
##### Annotating the HClust Dendrogram #####
##--------------------------------------- ##
# create an annotation dataframe
annotation.df <- data.frame(Cluster = as.factor(clusters))
rownames(annotation.df) <- rownames(Diagnostic.Features)
# apply a colour palette to the annotations on the dendrogram
# colour palette for the k clusters
k.cluster.colourpalette <- c("olivedrab2", "maroon3")
# colour mapping
colourmapping <- setNames(k.cluster.colourpalette, levels (annotation.df$Cluster))
# make a list of annotation colours
annotation.colours = list(Cluster = k.cluster.colourpalette)
# match the names of the annotation colours to the cluster levels ( 1-8)
names(annotation.colours$Cluster) <- levels(annotation.df$Cluster)
## ----------------------------------------- ##
##### visualising clustering in a heatmap #####
## ----------------------------------------- ##
# set a colour palette
# diverging - spectral
div.spectral.red.blue <- c("#4a100e", "#731331", "#a52747", "#c65154", "#e47961", "#f0a882", "#fad4ac", "#ffffff",
"#bce2cf", "#89c0c4", "#5793b9", "#397aa8", "#1c5796", "#163771", "#10194d" )
div.spectral.blue.red <- rev(div.spectral.red.blue)
# interpolate the colours for continuous scales
continuous.spectral.redblue <- colorRampPalette(div.spectral.red.blue) (256)
continuous.spectral.bluered <- colorRampPalette(div.spectral.blue.red)(256)
# set the breaks in the colour scale
palette.breaks <- seq(from= 0, to = 8, length.out = length (continuous.spectral.redblue) + 1)
# generate the heatmap
hclust.dissimilarity.heatmap <- pheatmap(dissimilarity.matrix,
cluster_rows = h.clusters,
cluster_cols = h.clusters,
annotation_row = annotation.df,
# annotation_col = annotation.df,
annotation_colors = annotation.colours,
color = continuous.spectral.redblue,
breaks = palette.breaks)
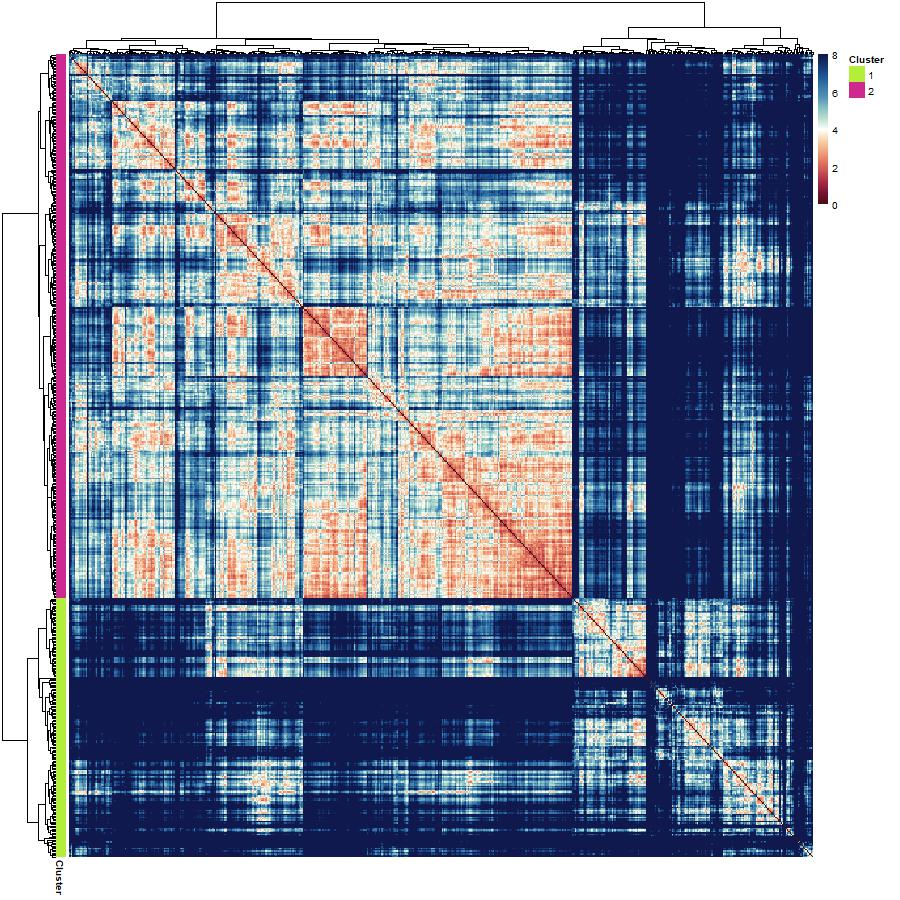
(6) Inspecter les résultats de votre regroupement hiérarchique :
Vous venez de produire un résultat de regroupement hiérarchique. Vous pouvez visualiser le regroupement à l’aide d’une carte thermique et d’un dendrogramme annoté, mais cela ne vous permet pas de savoir quelles cellules ont été reléguées dans quel regroupement. Vous pouvez également produire un diagramme à barres de proportions empilées qui vous indique la proportion de cellules bénignes et malignes dans un groupe.
Préparez un cadre de données de proportions – regroupez les données par groupe et par diagnostic et calculez les pourcentages.
préparez un diagramme à barres de proportions empilées à l’aide de ggplot et de l’élément geom_bar. D’après les résultats du regroupement, dans quelle mesure le regroupement hiérarchique a-t-il permis de distinguer les cellules bénignes des cellules malignes ?
– Question de l’étudiant – Parmi les caractéristiques de votre cadre de données, il existe des modèles qui permettent de distinguer les cellules bénignes des cellules malignes. Vous souhaitez évaluer la différence entre les cellules bénignes et les cellules malignes.
library(dplyr)
# Calculate proportions of benign and malignant cells by cluster
# you can use the pipe operator to nest a series of operations to be performed to the dataframe of origin
All.Samples.zscored.grouped <- All.Samples.zscored %>%
group_by(hclust_clusters, diagnosis) %>%
summarise(n = n()) %>%
mutate(freq = n / sum(n))
view (All.Samples.zscored.grouped)
# Prepare a stacked proportion barplot
Hclust.barplot <- ggplot(All.Samples.zscored.grouped, aes(fill=diagnosis, y=freq, x=hclust_clusters)) +
geom_bar(position="fill", stat="identity") +
theme_minimal() +
labs(x="Cluster", y="Proportion", fill="Diagnosis") +
scale_x_continuous(breaks = c(1,2))+
theme(axis.text.x = element_text(angle = 90, hjust = 1))
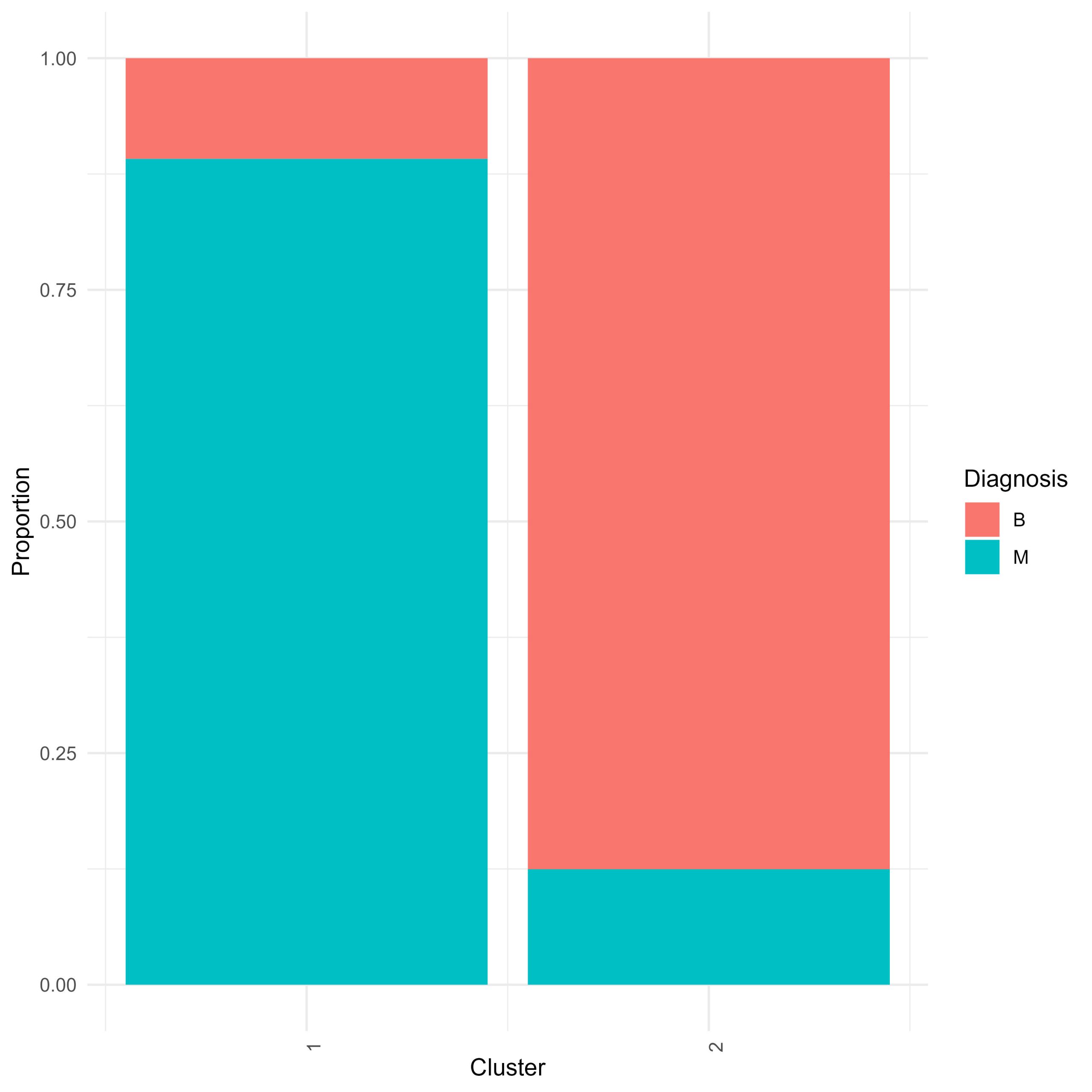
(7) Réalisation de statistiques d’estimation par étiquette de diagnostic
Les statistiques d’estimation constituent un cadre alternatif pour l’analyse statistique qui ne dépend pas des valeurs p ou des tests de signification. Au lieu de cela, elles montrent les données, les distributions et leurs différences médianes non appariées, ainsi que les intervalles de confiance à 95 % amorcées. Ce cadre vous permet de voir vos données et d’évaluer la différence ou l’absence de différence sans test de signification. Il s’agit d’un cadre puissant qui rend l’inférence statistique visuellement accessible et franchement belle !
(8) Réalisation des statistiques d’estimation par groupe hiérarchique
Les statistiques d’estimation précédentes ont été appliquées aux cellules classées par étiquette de diagnostic – pour évaluer les différences de caractéristiques entre les cellules bénignes et malignes. Cette fois, répétez le même cadre de statistiques d’estimation et examinez les mêmes caractéristiques, mais appliquez les statistiques d’estimation aux grappes à la place. Comment les graphiques d’estimation par grappe se comparent-ils aux graphiques d’estimation par étiquette diagnostique ?
Pour plus d’informations sur les statistiques d’estimation, veuillez consulter : Ho et al. 2019. publié dans Nature Methods
Ho, J., Tumkaya, T., Aryal, S. et al. Moving beyond P values : data analysis with estimation graphics. Nat Methods 16, 565-566 (2019). https://doi.org/10.1038/s41592-019-0470-3
# names of clusters
# cluster_names <- c("B", "M") # performing estimation stats by diagnostic label
cluster_names <- c("1", "2") # if performing estimation stats by hclust defined clusters
feature.of.interest <- "fractal_dimension_mean"
data1 <- All.Samples.zscored %>%
# select(variable = "diagnosis", value = feature.of.interest) # estimation on diagnostic labels
select (variable = "hclust_clusters", value = feature.of.interest) # estimation on the hclust clusters
estimation.stats.data <- data1
# Specify your reference group
# reference_group <- "B" # estimation by diagnostic label ( B = Benign, M = Malignant)
reference_group <- "1" # estimation by hclust cluster
# set the control group
control = reference_group
# set the comparison groups
comparisons = setdiff(cluster_names, control)
# Set the column names
colnames(estimation.stats.data)[2] = "Z-Scored Mean Nuclear Fractal Dimension"
# Prepare data for estimations statistics processing
two.group.unpaired =
estimation.stats.data %>%
dabest(variable,`Z-Scored Mean Nuclear Fractal Dimension`,
idx = c(control, comparisons),
paired = FALSE) %>%
median_diff(reps = 10000)
# Set the color parameters
# colour palette corresponds to diagnostic labels
# colours = c("coral2", "turquoise3" )
# colour palette corresponds to hierarchically defined clusters
colours = c("maroon3", "olivedrab2")
colour.swarm.plot = c(colours[which(cluster_names == control)],
setdiff(colours,
colours[which(cluster_names == control)]))
swarm.plot <- plot(two.group.unpaired,
palette = colour.swarm.plot,
# tick.fontsize = 20,
# axes.title.fontsize = 25,
rawplot.type = "swarmplot",
rawplot.ylabel = "Z-Scored Mean Nuclear Fractal Dimension")+
ggtitle(paste("Reference: cluster ", control))+
theme(title = element_text(face = "bold"),
plot.title = element_text(hjust = 0.5, vjust = 10, size = 20,
margin = margin(t = 80, b = -35)))
print(swarm.plot)
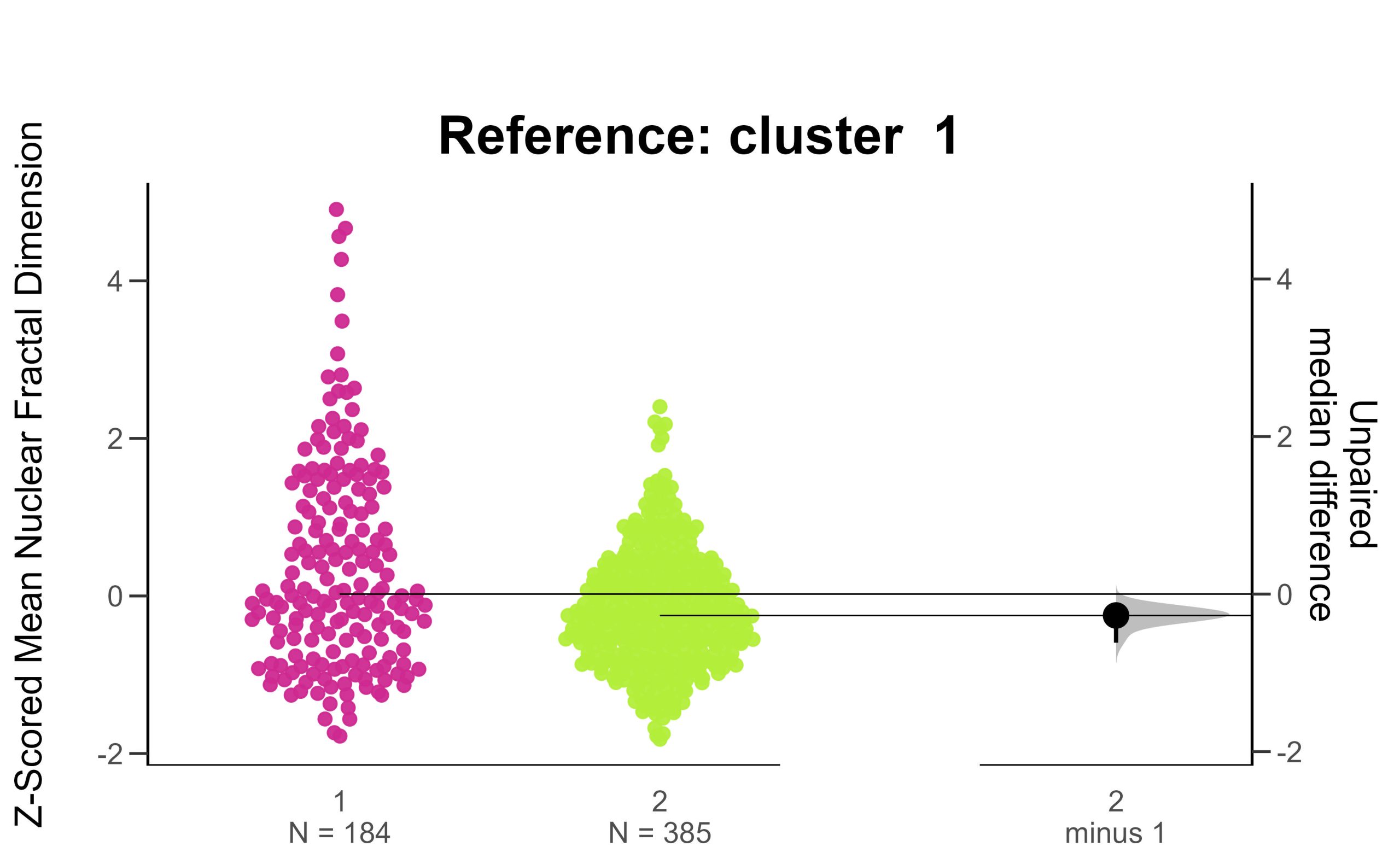
(9) Comparer le regroupement hiérarchique aux étiquettes de diagnostic avec la réduction de la dimensionnalité
Une autre façon d’évaluer l’efficacité de l’affectation des grappes en fonction de l’étiquette de diagnostic consiste à annoter le graphique T-SNE que vous avez construit précédemment, mais cette fois-ci, au lieu de colorer les points en fonction de leur étiquette de diagnostic connue, vous pouvez les annoter en fonction de leurs grappes définies de façon hiérarchique.
Utilisez la même perplexité et la même graine aléatoire que précédemment. De cette façon, vous pouvez comparer la façon dont les grappes de grappes sont représentées dans l’espace à haute dimension et la façon dont les étiquettes de diagnostic sont représentées dans le même espace. Lorsque vous comparez les graphiques t-SNE annotés par diagnostic et par grappe hiérarchique, que remarquez-vous ? Quelles sont les similitudes et les différences qui vous semblent évidentes ?
## --------------------------------------------------- ##
##### Repeat T-SNE code but plot coloured by hclust #####
## --------------------------------------------------- ##
# Set your perplexity and seed
set.seed (789)
perplexity <- 30
seed <- 789
# Perform t-SNE with the specified perplexity and seed
tsne_model <- Rtsne(Diagnostic.Features.Scored, perplexity = perplexity, seed = seed)
# Create a dataframe for the t-SNE components
tsne_data <- data.frame(
tSNE1 = tsne_model$Y[, 1],
tSNE2 = tsne_model$Y[, 2],
hclust_cluster = as.factor(All.Samples.zscored$hclust_clusters) # Add the Hierarchical Cluster Assignment - convert to factor to make the scale discrete
)
view( tsne_data)
# Load ggplot2
library(ggplot2)
# specify the colour palette :
hclust.colours <- c("maroon3", "olivedrab2")
# Create the plot
tsne.by.hcluster <- ggplot(tsne_data, aes(x = tSNE1, y = tSNE2, colour = hclust_cluster)) +
geom_point() +
labs( x = "T-SNE Component 1", y= "T-SNE Component 2") +
theme_bw() +
scale_color_manual(values = hclust.colours)
# Print the plot
print(tsne.by.hcluster)
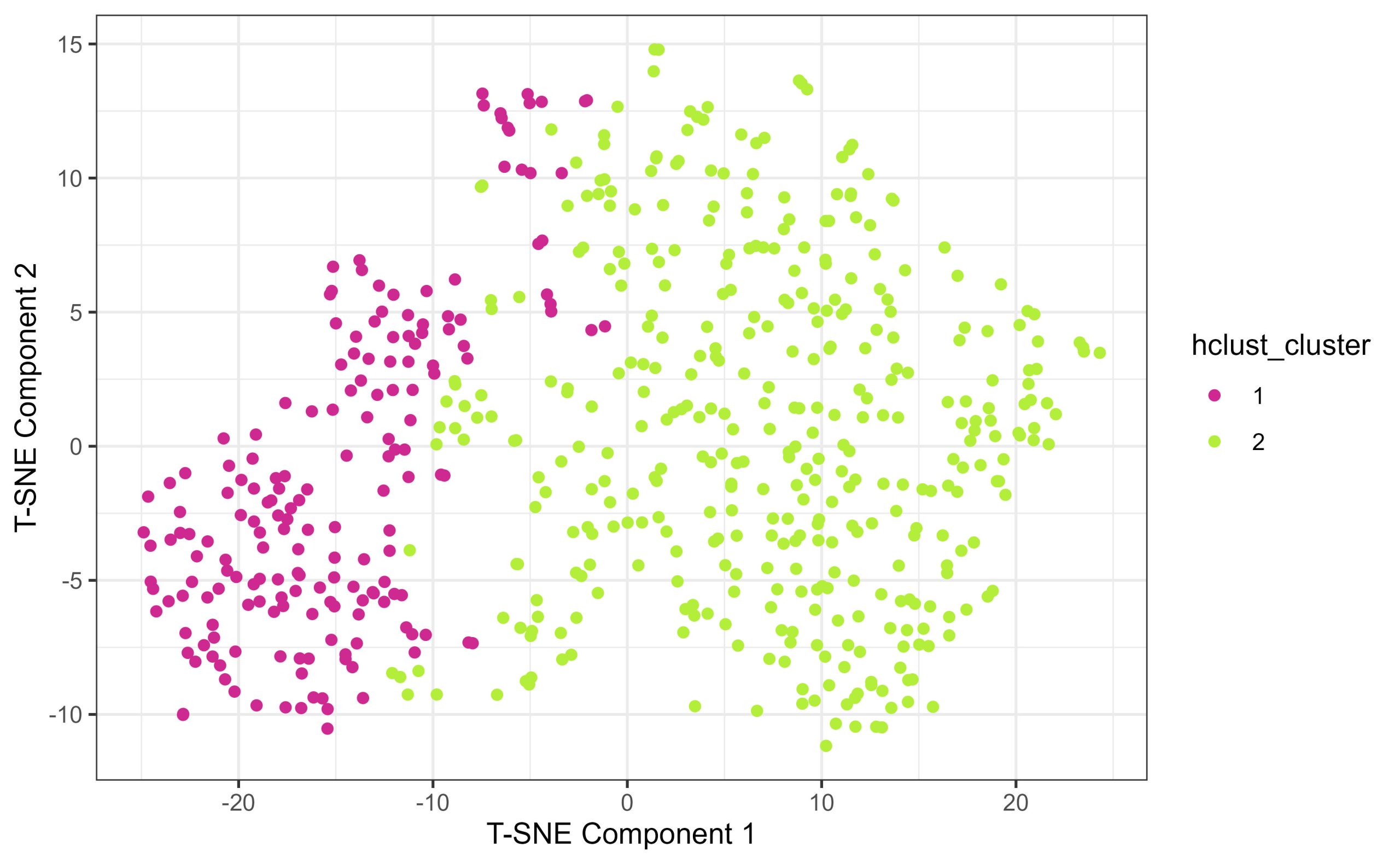
(10) Quelques derniers éléments de réflexion
(réflexion des élèves – peut donner lieu à une discussion sur les algorithmes de regroupement et sur les raisons pour lesquelles il est important de vérifier et de valider les résultats des regroupements)
Vous avez comparé les véritables étiquettes diagnostiques des cellules cancéreuses du sein – bénignes et malignes – à des clusters hiérarchiques construits uniquement à partir de caractéristiques de taille et de forme. Compte tenu de l’efficacité avec laquelle l’algorithme de regroupement a séparé les cellules bénignes et malignes, réfléchissez à l’utilité de cette approche si vous ne saviez pas a priori que les cellules étaient classées dans les catégories de diagnostic bénigne et maligne. Après les avoir regroupées en deux groupes et avoir comparé leurs statistiques, que devriez-vous faire pour confirmer que les cellules d’un groupe sont malignes et que les autres sont bénignes ?
