5 Drug Treatments for Pain and Inflammation
5.1 Pathology
Responding to tissue damage is usually accompanied by a physiological and psychological response via inflammation and pain. Although these are associated with negative responses, acute pain and inflammation are important in helping remove the negative stimuli, fighting off foreign invaders, and healing injuries. However, prolonged inflammation and pain can lead to additional damage and discomfort. The regimen of drugs developed for pain and inflammation are designed to deal with physiological and/or psychological responses.
In the simplest example, damage to a cell can occur through a puncture or rupture of the membrane which can lead to a cascade of different responses. For example, one initial aspect can involve the influx of Ca2+ into the cell. Ca2+ is a potent biochemical messenger and multiple proteins and transcription factors are sensitive to Ca2+ binding which can trigger different responses depending on the type of cell. One of the key responses is the activation of a protein called phospholipase A2 (PLA2). PLA2 interacts with phospholipids of the cell bilayer and will cleave fatty acids from the phospholipid.
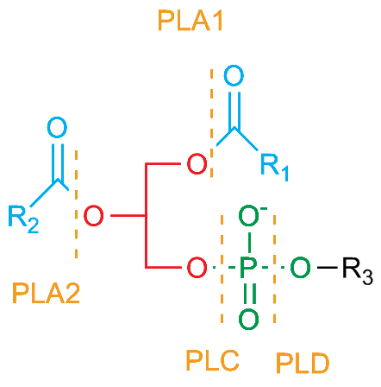
Recall that the structure of a phospholipid has a glycerol backbone with a phospholipid head group esterified to one oxygen of glycerol (called stereospecific numbering 1 or SN1) and fatty acid chains esterified to the other two oxygen atoms (called SN2 and SN3). (Figure 5.1) Phospholipase enzymes cleave at specific SN positions (PLA1 cleaves at the SN1 position, PLA-2 cleaves at SN2, PLAB cleaves at either SN1 or SN2, and PLAC and PLCD cleave on specific sides of the phosphate at the SN3 position). During the inflammatory responses, one of the most important phospholipases is PLA2 which cleaves the fatty acid in the middle (SN2) position. The fatty acid released is often an arachidonic acid. (Figure 5.2)
5.1.1 Arachidonic acid and inflammation
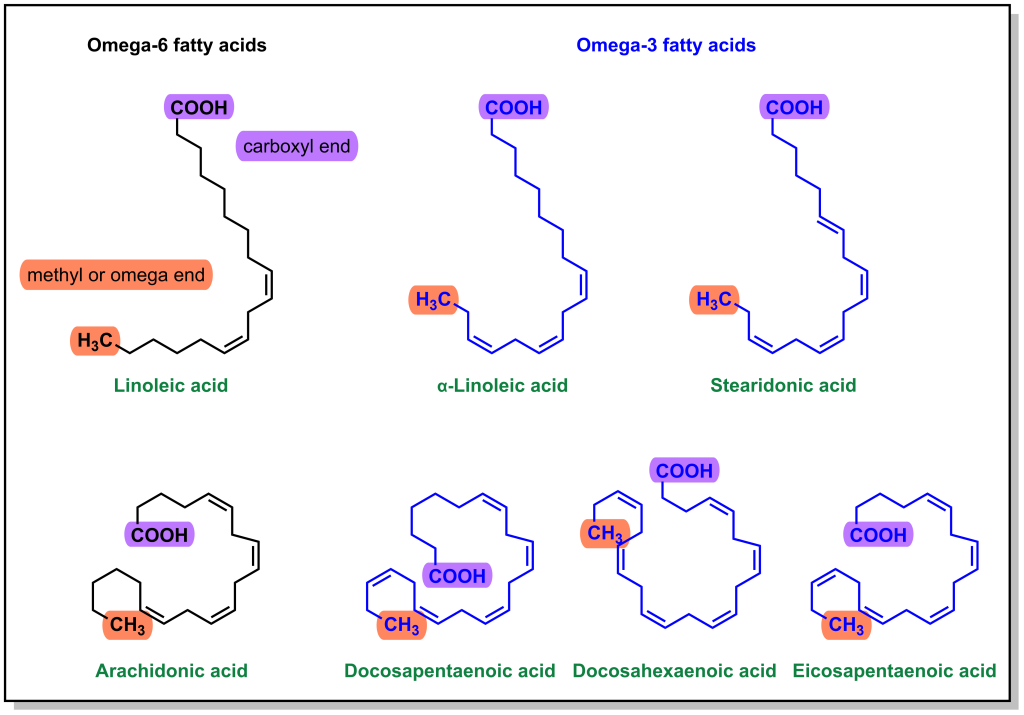
There are multiple types of fatty acids that are incorporated into phospholipids. Since physiological fatty acids can have variable lengths and degrees of saturation, historically it has been more convenient to name fatty acids from the omega end (final carbon) rather than the alpha end. Omega-3 fatty acids have a double bond at the 3-C from the omega position and omega-6 fatty acids have a double bond at the 6-C from the omega position. (Figure 5.2) Arachidonic acid is an omega-6 fatty acid and it contains 4 double bonds, which are in the ‘cis’ configuration, leading to a more closed geometry that contributes to cell membrane fluidity. The double bonds are also highly susceptible to oxidation, which makes arachidonic acid a good target for downstream signalling pathways. (Figure 5.3)
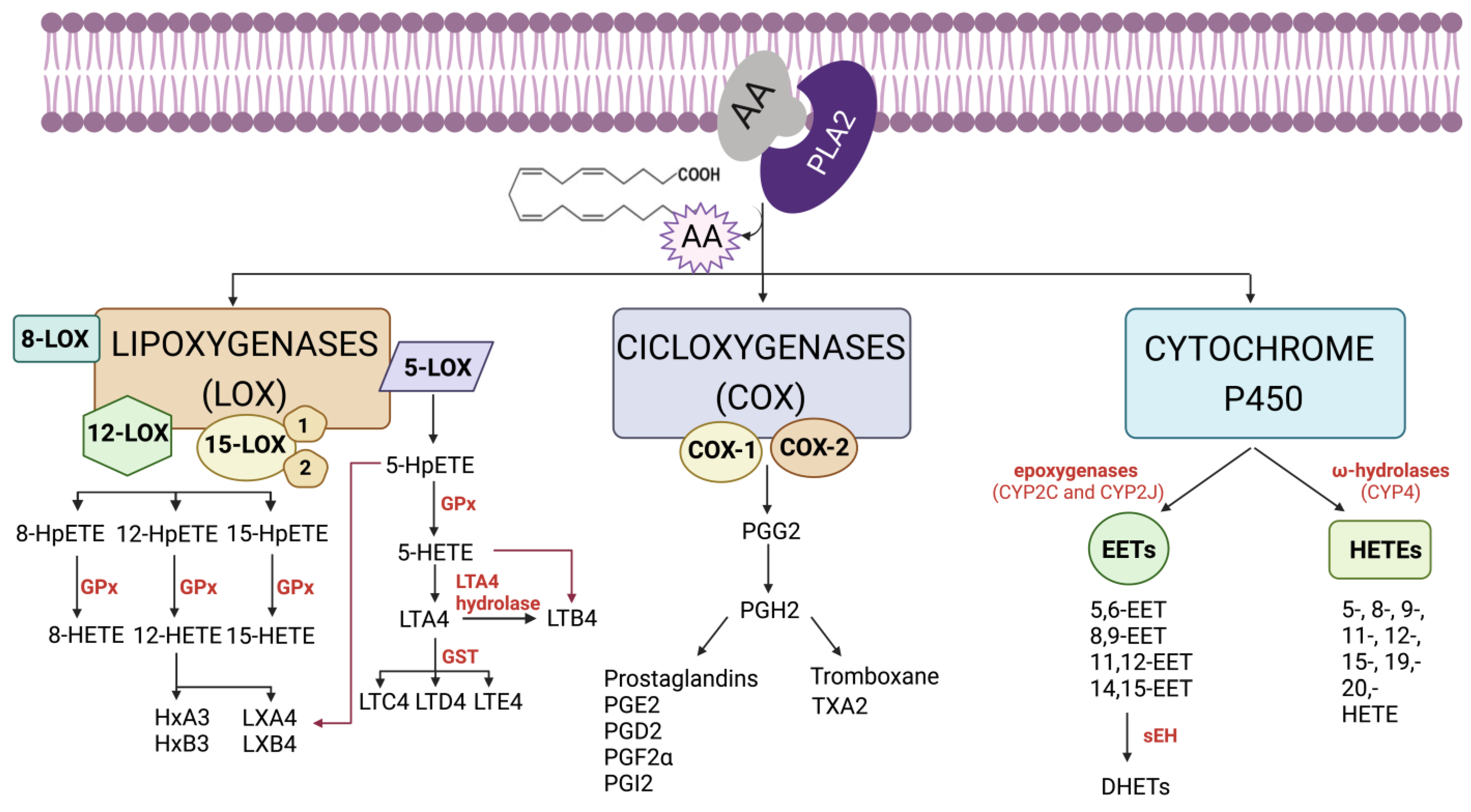
Arachidonic acid signalling pathways begin following the influx of Ca2+ and activation of PLA2 which cleaves specific phospholipids and releases arachidonic acid from the SN2 position. Free arachidonic acid acts a substrate for many different proteins, which are mainly stratified into three classes, cyclooxygenases (COX enzymes), cytochrome P450 (CYP) enzymes, and lipoxygenases (LO enzymes). Arachidonic acid is oxidized into different molecules that are called eicosanoids and includes molecules such as prostaglandins and thromboxanes. Each of these molecules can have different effects, such as increased inflammation or platelet aggregation.
Inhibition of this pathway can occur at different points. At the top of the pathway is PLA2, and this enzyme is potently inhibited by steroids, which shuts down the inflammatory response. However, steroids are hormones, and they can have multiple effects throughout the body. Therefore, targeting enzymes lower in the biochemical response pathway such as arachidonic acid metabolizers represents a more selective strategy. The most common approach is inhibition of the COX enzymes which generate the prostacyclins, prostaglandins, and/or thromboxanes.
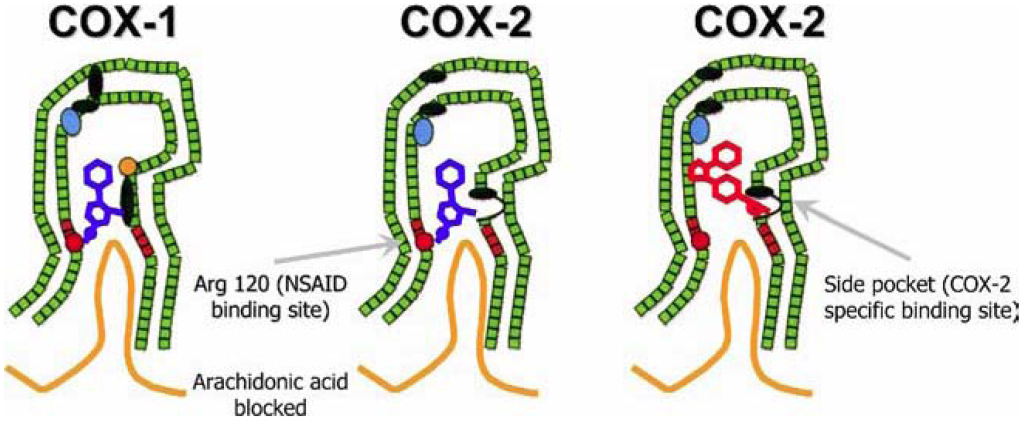
There are two key isoforms of the COX enzymes, referred to as COX1 and COX2. COX1 is shown to be constitutively active, albeit at low levels in multiple tissues, and helps maintain homeostasis. COX1 has important roles in maintaining the stomach lining, normal functioning of the kidneys, and platelet aggregation. COX2 is upregulated in response to inflammatory signals. Upon activation, both COX1 and COX2 are found at the cell membrane surface and are positioned in such a way that a long channel within the enzyme is oriented towards the membrane to accommodate any free arachidonic acid molecules. (Figure 5.4)
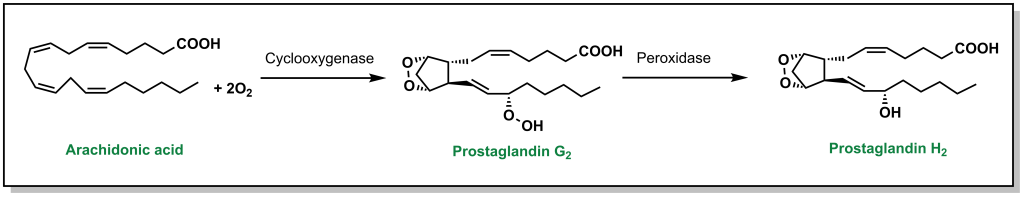
When arachidonic acid is bound inside the COX enzymes, it is oxidized to form prostaglandin G2, which is converted into prostaglandin H2 and serves as a substrate for other enzymes. (Figure 5.5)
5.2 Treatments for Pain and Inflammation
Since the molecules involved in targeting the COX enzymes are not steroids but still reduce inflammation, they are referred to as Non-Steroidal Anti-Inflammatory Drugs or NSAIDs. Although the COX enzyme was first isolated and purified in 1976, it has been inhibited by the action of natural products used by multiple civilizations (such as the bark of white willow or meadowsweet trees). The active ingredient from these plants (salicylic acid) was eventually identified, and some of the more adverse reactions (such as GI discomfort) were ablated with the use of the esterified acetylsalicylic acid (aspirin). Aspirin was the first drug to be developed, patented, and marketed in 1899 and represents a large milestone in the history of pharmaceutical agents.
5.2.1 Aspirin
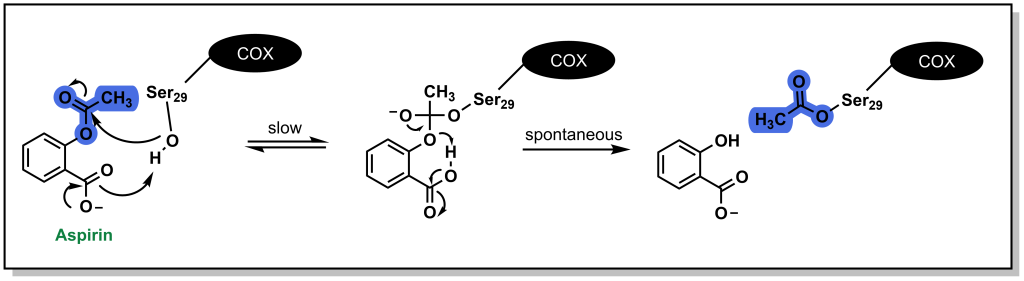
The anti-inflammatory effects of aspirin arise through inhibition of the COX enzyme. Acetylsalicylic acid binds the positively charged side chain of Arg120 on the COX enzyme and blocks arachidonic acid from entering the protein channel and being oxidized to inflammatory metabolites. (Figure 5.4) Aspirin also has a unique mechanism of action in which it covalently (or irreversibly) engages with the COX enzyme by reacting with Ser29. (Figure 5.6) The acetyl-group acts a leaving group, and the COX enzyme is permanently modified with the salicylic acid. Cells (such as those in the gastric lining) need to biosynthesize new COX1/COX2 enzymes to restore proper function. However, unnucleated cells (such as platelets) cannot re-synthesize these proteins and aspirin is a potent inhibitor of platelet aggregation.
5.2.2 Other Non-steroidal Anti inflammatory Drugs
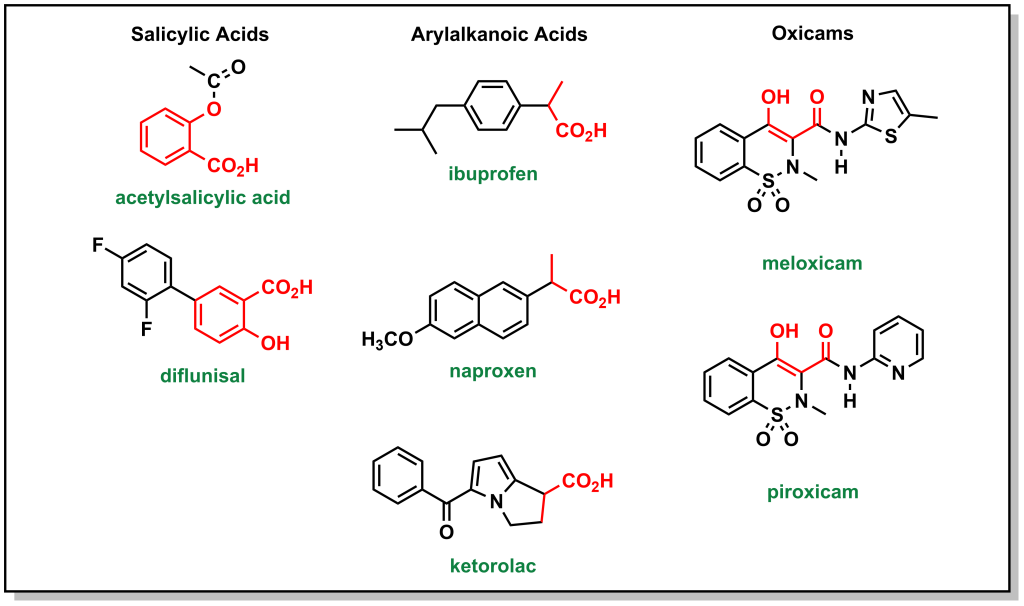
There are multiple NSAIDs that are employed (other than aspirin), although they differ in that they do not operate via a covalent mechanism of action. They can generally be classified into salicylic acids, arylalkanoic acids, and oxicams. (Figure 5.7) In all cases, they have an acidic functionality that is critical to engage with Arg120 of the COX enzyme and sterically block access to the pocket.
5.2.3 Tylenol
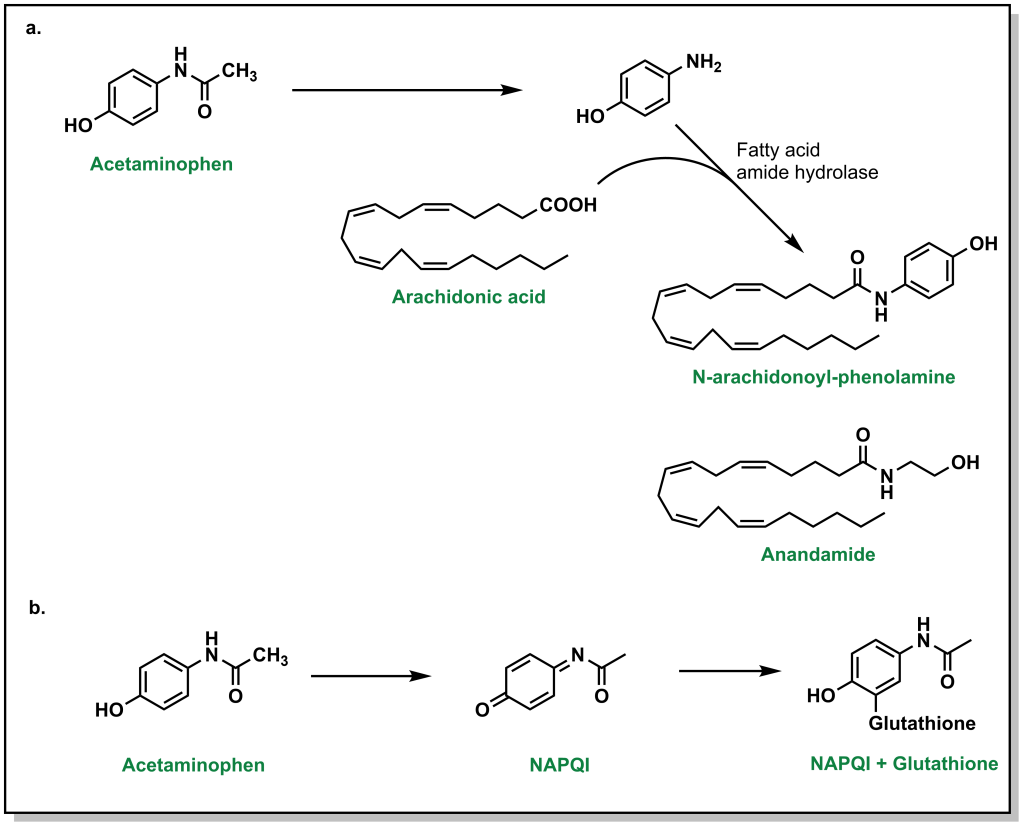
Tylenol or acetaminophen is a common analgesic that was previously thought to inhibit COX enzymes. However, it was shown to only weakly bind to the COX enzymes in vitro and as such is not an NSAID, likely due to low acidity of the hydroxyl group. Acetaminophen is thought to act centrally in the brain to block pain signals and directly engage with arachidonic acid. (Figure 5.8a) However, it is also capable of inducing anti-inflammatory responses. One of the largest concerns with Tylenol is the potential for toxicity with excessive doses. Nearly all (~95%) Tylenol is metabolized by glucuronidation and sulfonylation. (Figure 5.8b) However, a small amount will be metabolized by CYP2E1 which leads to generation of the superoxide and NAPQI (N-acetyl-p-benzoquinone imine). This intermediate is toxic because it is highly reactive and can covalently modify different residues or nucleotides. In the presence of glutathione, NAPQI is rapidly de-toxified. However, if there are substantial amounts of Tylenol, this will deplete the reservoir of liver glutathione leaving free NAPQI available to react with other biomolecules.
5.2.4 Opioids
Pain generally begins at the nociceptors, and the signal is transmitted to secondary neurons in the dorsal root ganglion and subsequently to the brain via the spinal cord nerves. The signal transmission occurs via action potentials that fire with increased frequency depending on the severity of the pain. Neurons release multiple neurotransmitters that help facilitate different responses:
- Glutamate: This activates different receptors on the post-synaptic neuron (such as NMDA, AMPA) and causes Ca2+ and Na+ to enter the cell (increasing the positive charge of the cell)
- Substance P: This leads to the release of substances of such as arachidonic acid, and reinforces the pain signal.
- CGRP: This alters GPCR expression which reinforces response to pain.
At the same time, the body also releases endogenous molecules to help alleviate some of the effects of pain such as dynorphins and endorphins. These molecules can bind to mu, delta, or kappa receptors. Euphoric effects arise upon binding and activation of the mu receptors in the brain. These receptors reduce the response of GABA receptors, and the net result is the removal of limiters on dopamine release. These natural ligands produce analgesic effects. However, opioids are substantially more potent binders and lead to more powerful analgesic effects.
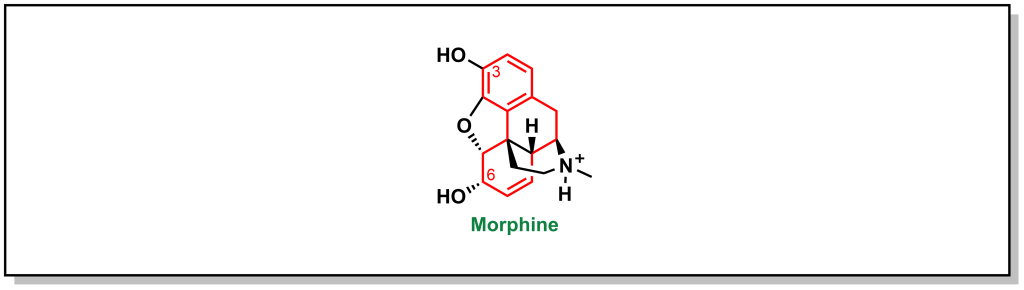
Opioids have been used for thousands of years and they originate from the milky fluid (called latex) of the poppy plant. Morphine is the primary alkaloid of the opium family. (Figure 5.9) It contains multiple functional groups that are critical for analgesic activity. This includes:
- A tertiary nitrogen that is positively charged to engage with the side chain of an aspartic acid on the mu receptor.
- The tertiary nitrogen attached to a quaternary carbon by a 2-C bridge.
- The quaternary carbon has a phenyl group attached for pi-pi stacking interactions with the mu receptor.
- Functional groups on C3 and C6 are important for H-bonding interactions.
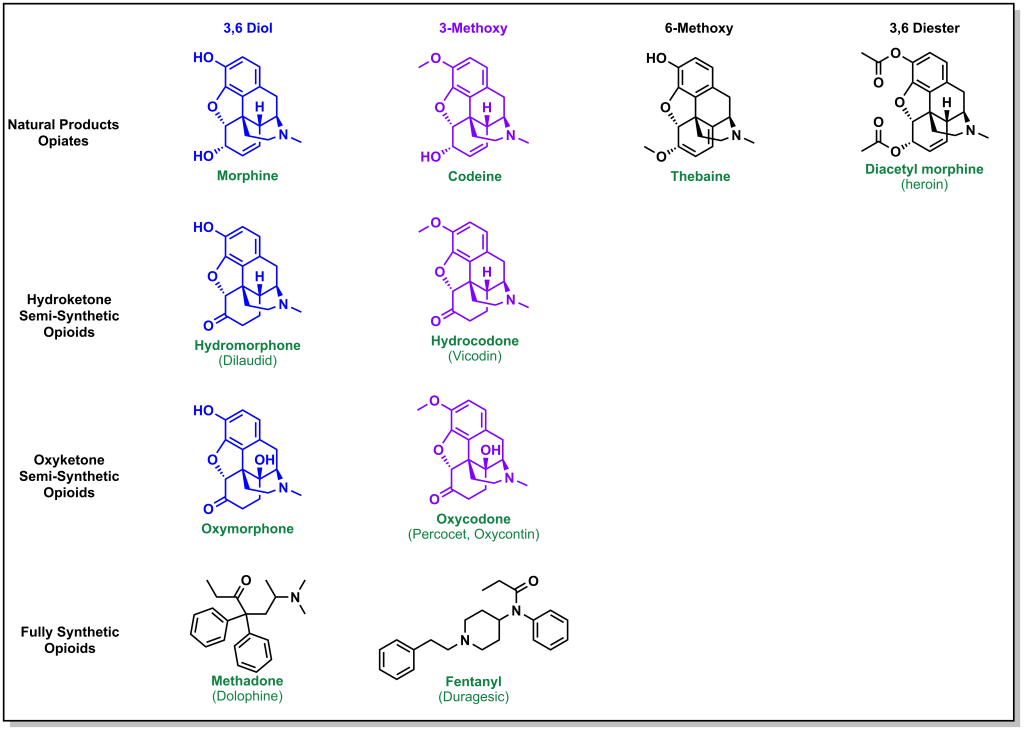
Natural, synthetic, and semi-synthetic opioids have been explored which offer alternative properties in bioavailability. (Figure 5.10) For example, codeine is a naturally occurring opiate, and the only difference between morphine, is in the C3 position (with a methoxy instead of the hydroxyl). This increases the lipophilicity of the molecule substantially. However, the C3 methoxy can no longer participate in H-bonding and also has substantially lower binding to the mu receptor. Although codeine would be extremely potent if it was metabolized to morphine, this does not occur efficiently. In fact, even though the lipophilicity of codeine is higher than morphine, because of the slow rate of metabolism, an oral dose of 100 mg is required to achieve the same response as 10 mg of morphine.
Heroin is a version of morphine where the hydroxyl groups on C3 and C6 are both acetylated. This increases the lipid/membrane solubility leading to rapid CNS penetration. The acetyl groups are also readily metabolized to hydroxyls (unlike codeine) and heroin is approximately two-fold more potent than morphine.
To compensate for the low binding affinity of codeine, different derivatives were examined such as hydrocodone. In hydrocodone, the hydroxyl at C6 is converted to a ketone which increases the lipophilicity while preserving the double bond. However, the carbonyl alters the positions of the oxygen relative to the hydrogen bond donor and removing the double bond at C3-C4 helps re-align the atoms. These changes result in similar potencies between hydrocodone and morphine. Hydrocodone can be metabolized to more potent hydromorphone which is similar with the exception of the hydroxyl instead of methoxy at C6.
Another SAR modification was the introduction of a hydroxy at C14 to hydrocodone. Oxycodone has further improved bioavailability and is about two-fold more potent than morphine.
5.3 Summary
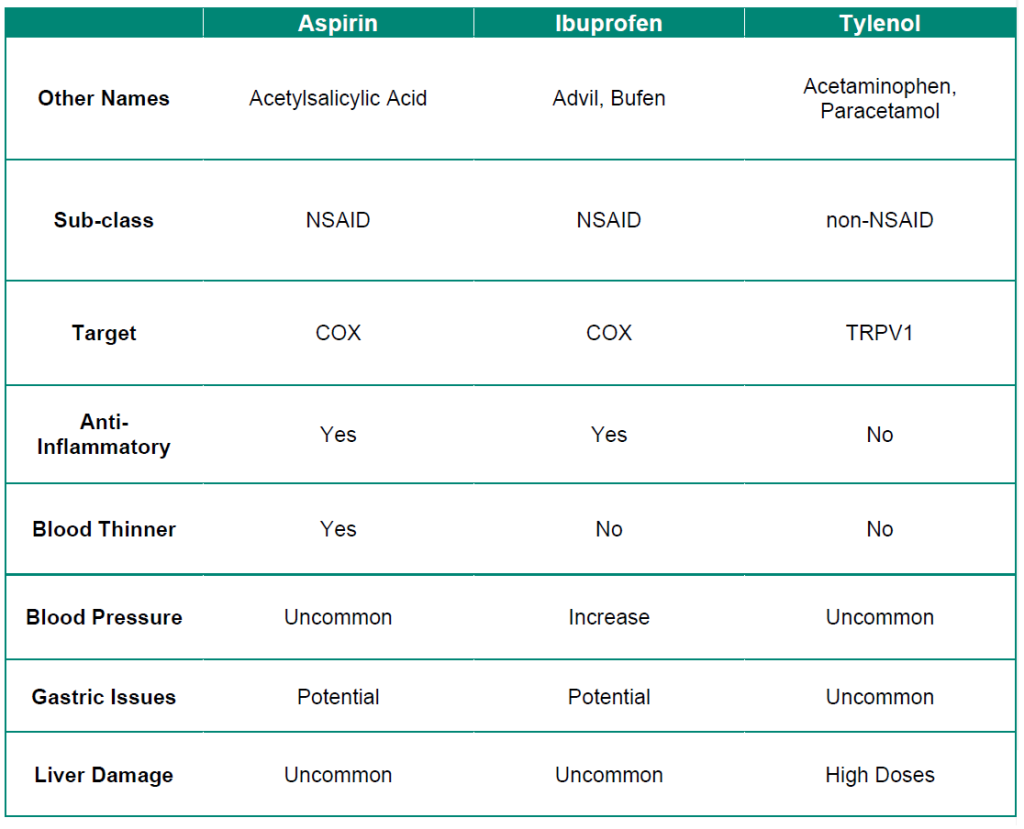
Table 5.1: Summary of over-the-counter analgesic drugs.

{kind=link}