2 Drugs for Treatment of Venous Thromboembolism
2.1 Pathology
The circulatory system distributes blood throughout the body, carrying oxygen, nutrients, and hormones to different organs, while removing carbon dioxide and other waste products. Blood contains multiple components including blood cells and an array of different proteins, lipids, and carbohydrates. Maintaining a constant flow of blood is essential for viability of all organs and tissues. Venous thromboembolism (VTE) refers to a blockage that occurs from a blood clot in the veins and impedes blood flow. There are generally two types of VTE, which refer to the location the blockage – deep vein thrombosis (DVT) or superficial vein thrombosis (SVT). Superficial veins are closer to the surface of the skin and are often translucently visible. Blood clots that occur in these regions usually resolve naturally (on the timescale of hours to days) and are less medically serious unless they travel to the deep veins (such as within the legs). Conversely, DVT can lead to life-threatening conditions including pulmonary embolisms and must be treated as soon as possible.
2.1.1 Blood Flow Blockages
Blockages that occur in the blood have different names. If blood cells aggregate and form a semi-solid mass that is attached to a blood vessel, this is referred to as a thrombus. If this thrombus detaches from the blood vessel, it is referred to as a blood clot. This is a minor but significant difference. For example, if blood is left in a test tube and coagulates into a semi-solid mass, it would be called a blood clot (and not a thrombus) because it is not attached to any blood vessels. A blood clot is also a specific example of an embolus, which is a substance that travels through the blood stream and can create a blockage. Blood clots, gas bubbles, cholesterol aggregates, or foreign bodies can all be identified as emboli within the blood stream.
There are situations in which blockage of blood flow is required, especially when there is damage to the circulatory system and bleeding occurs. In this case, the body has a system of responses to limit bleeding and repair the damage. This occurs in two stages called primary hemostasis, where platelets assemble in the area of damage and start to stick to each other, and secondary hemostasis (or coagulation), where a mesh of proteins (mostly a protein called fibrin) forms to hold the platelets together. During coagulation, the strands of fibrin protein wrap around the platelet plug that forms during primary hemostasis and become insoluble (via the action of FXIIIa), helping block the loss of blood.
2.1.2 Coagulation Cascade
Coagulation or the clotting of blood is controlled by “clotting factors” or “coagulation factors” in the biochemical pathway known as the coagulation cascade. In total, there are 12 clotting factors labelled with Roman numerals (I, II, III, IV, V, VII, VIII, IX, X, XI, XII, and XIII). Note that there is no clotting factor VI, since these proteins were numbered in the order of their discovery, and the protein species identified as clotting factor VI was later recognized as a different version of clotting factor V. Importantly, the clotting factors exist in two forms, an active form and an inactive form. The active form is labelled with a lower case ‘a’ following the Roman numeral. In addition to this naming convention, specific active and inactive factors have common names, which are identified in Figure 2.1.
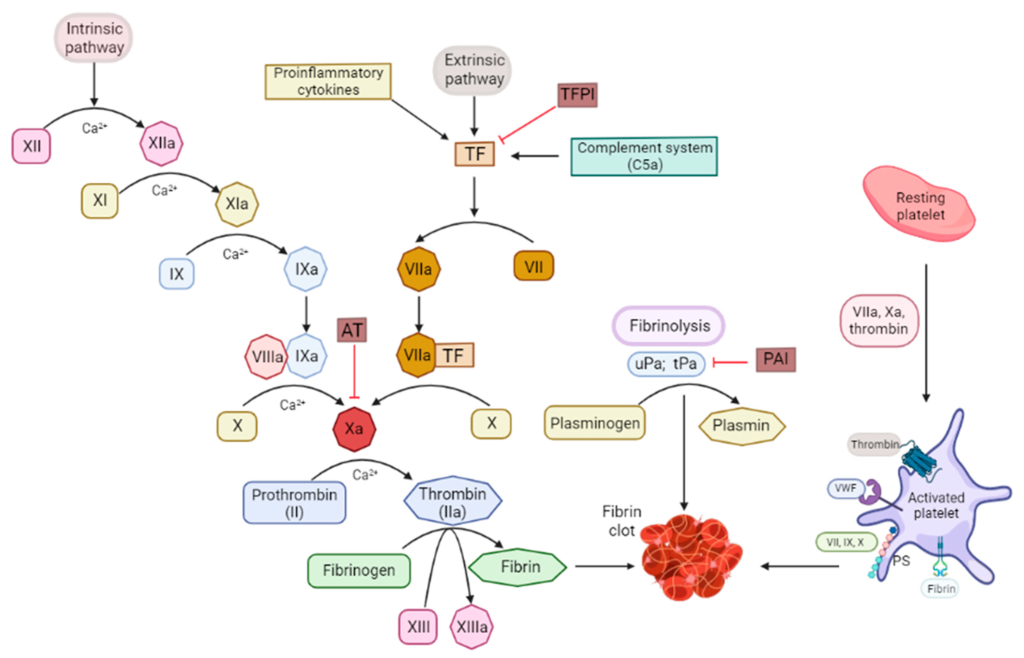
These factors all participate in the coagulation cascade, which has an end-goal of creating fibrin protein (factor Ia) for the insoluble mesh formation. Several of these factors are enzymes with serine protease activity that will cleave and activate the downstream factor. The biochemical underpinnings of the full cascade are complex and can be initiated when there is internal vascular endothelium damage (intrinsic pathway) or external trauma (extrinsic pathway). Regardless, both pathways converge of activation of factor X to Xa, which leads to activation of thrombin (IIa) and fibrin (Ia). This pathway is also naturally regulated by the protein anti-thrombin, which can bind thrombin (IIa), but also has activity for blocking factor Xa.
The goal for anti-coagulation treatments is to interfere with any of these processes in the coagulation cascade, which will effectively enable shutting down the fibrin formation process and forms the mechanism for all anti-coagulants that are currently on the market.
2.2 Anti-Coagulant Treatments
2.2.1 Warfarin
Warfarin is an anti-coagulant that was approved in 1954, and there is a substantial amount of clinical information available on its use across many patients. As such, warfarin is the most clinically comfortable anti-coagulant to prescribe. Warfarin targets factor X and II (Pro-thrombin) but also blocks factor IX and VII, leading to very potent anti-coagulation effects. These broad inhibition effects arise from the Ca2+-binding requirement of these enzymes. Warfarin blocks the proper synthesis of these factors and prevents them from binding Ca2+ in an intricate pathway.
2.2.1.1 Warfarin Blocks VKORC Leading to Downstream Effects on the Clotting Factors
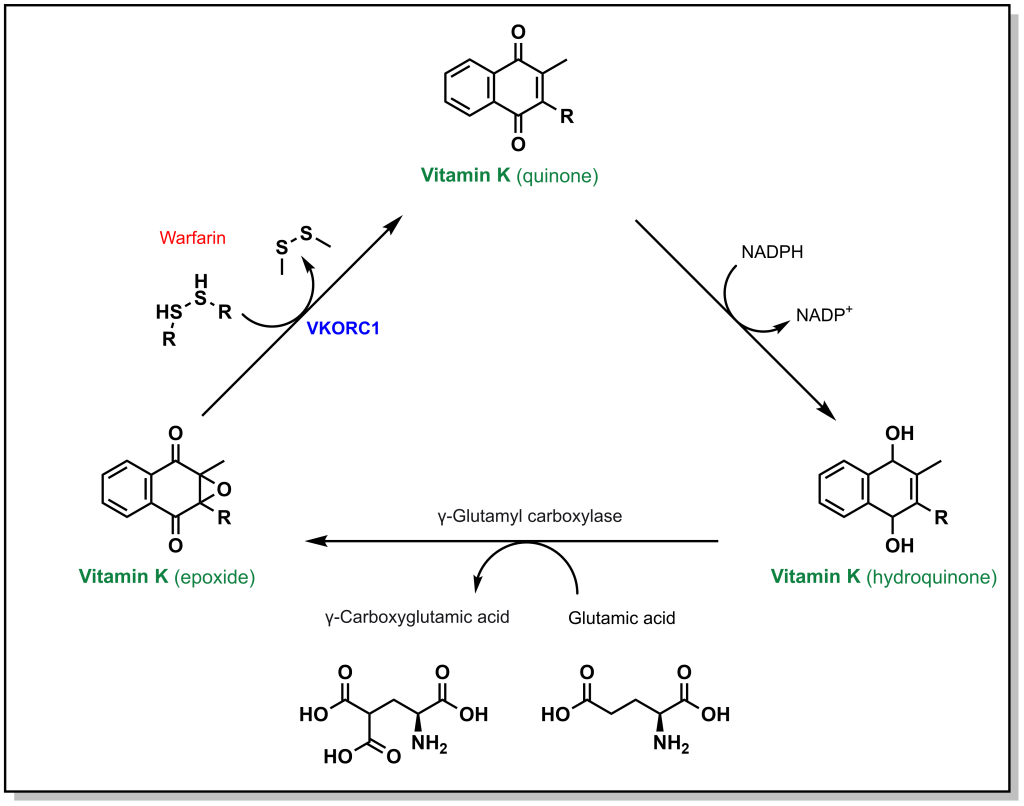
Calcium ion binding within blood clotting factors occurs in a unique binding mode. The blood clotting factors X, II, IX, and VII all possess a specific glutamic acid where the side chain is converted to a di-carboxylic acid. Unlike normal glutamic acid side chains where there is a single acid functionality, this modified amino acid now possesses two carboxylic acids that enables it to chelate a Ca2+ ion. The di-carboxylation is performed by the enzyme gamma glutamyl carboxylase in a vitamin K dependent process. In this process, Vitamin K is converted from a hydroquinone to an epoxide resulting in an inactive Vitamin K species. In order to generate an additional di-carboxylated glutamic acid, Vitamin K must also be re-generated. The re-generation is carried out an enzyme called vitamin K epoxide reductase complex 1 (VKORC1). Warfarin can competitively bind at the active site of VKORC1 and block this reaction, thereby blocking the regeneration of vitamin K. (Figure 2.2)
2.2.1.2 Properties of Warfarin
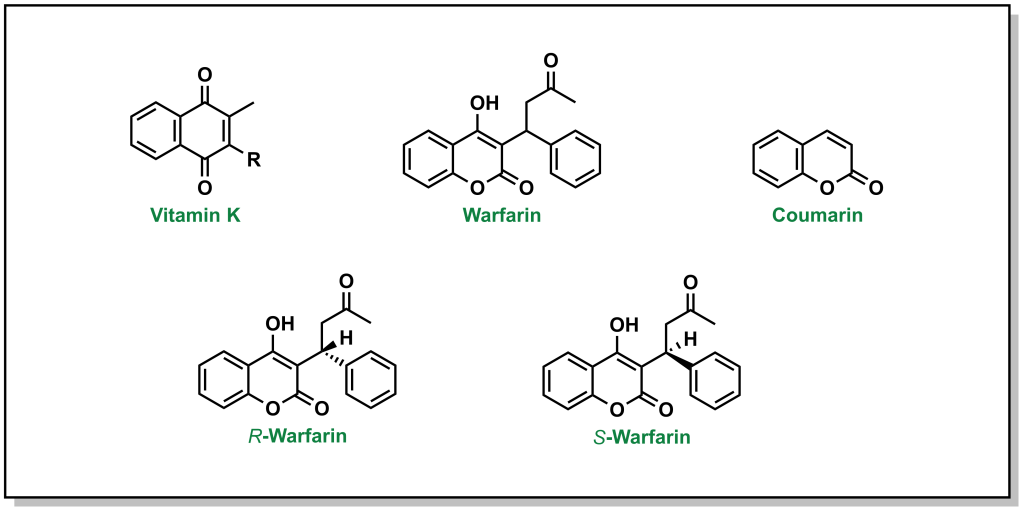
Vitamin K is a quinone (6-membered unsaturated ring that is conjugated with two carbonyls). There are two versions of vitamin K found in humans, which depend on the saturation level of the hydrophobic tail. Warfarin will competitively displace vitamin K from the active site of VKORC1. Unlike vitamin K, warfarin has a coumarin structure. (Figure 2.3). Importantly, warfarin possesses a stereogenic centre leading to two different chiral molecules (R-warfarin and S-warfarin). The S– enantiomer is ~5-fold more active, although the pharmaceutical drug is delivered as a racemic mixture (equal concentrations of both enantiomers).
Warfarin can be administered orally and displays a bioavailability of >99%, which indicates that nearly all the drug ingested will be absorbed and enter the blood stream without degradation/metabolism (which is unusual for an oral drug). As such, warfarin also has a relatively long half-life in the body (>40 h). However, it is also a slow-acting drug in that it will take ~3-4 days to observe any effects. This is because the mechanism of action of warfarin involves blocking synthesis of new blood clotting factors, and it will not have an effect on the current proteins. Therefore, the “older” blood clotting factors need to complete their normal lifecycle in the body and be degraded before the effects of warfarin are observed (blood clotting factor turnover usually occurs within 3-4 days). Warfarin is also teratogenic and crosses the placenta and should not be used if the patient is pregnant.
Warfarin dosing can also be a complicated process, since there can be a high degree of variability between different individuals – by 20-fold in extreme cases. From a dietary perspective, since warfarin is a competitive vitamin K inhibitor, if patients consume foods with significant quantities of vitamin K (i.e. leafy green vegetables) this can reduce the effects of warfarin. For these reasons, patients are often recommended to maintain a consistent level of leafy greens in their diet to avoid continuous variations in dosing. Another point of variability is that warfarin is metabolized by the CYP2CP enzyme, and specific genetic populations have different isoforms that metabolize warfarin at different rates. For example, CYP2C9*2 (R144C variant) and CYP2C9*3 (I359L variant) lead to significant reduction in metabolic activity. Based on all of these factors, warfarin dosing needs to be monitored closely and adjusted (via an INR [international normalized ratio] test, which provides a quantitative measure of how fast blood clots from a patient).
2.2.2 Heparins
After warfarin, heparins emerged as the second most common anti-coagulant. While warfarin is a small molecule inhibitor that does not occur naturally in the body, heparin is a naturally occurring sugar derivative. The structure of heparin involves a specific sequence of five carbohydrate monomers (pentasaccharide sequence) followed by additional carbohydrate monomers of variable length. Naturally occurring heparin is a collection of these sugars with different sizes/lengths of sugar tails.
The main target for heparin is the protein anti-thrombin. Recall that anti-thrombin regulates the activity of both thrombin (factor IIa) and also factor Xa. (Figure 2.1) This is carried out by the conserved pentasaccharide sequence found on all heparin molecules that binds to anti-thrombin. This binding triggers a conformational change at the active site of anti-thrombin which enables it to engage with either factor Xa or factor IIa (since the binding sites are similar on both proteins). (Figure 2.4) When anti-thrombin binds to thrombin (factor IIa), the heparin can wrap around anti-thrombin (in a process called ‘bridging’) and engage the bound thrombin protein. This bridging function blocks the protein more effectively (provided it is long enough – i.e. heparin has at least 13 sugar molecules). This second bridging interaction is generally required for heparin to fully block the activity of thrombin. However, this interaction site does not occur between heparin and factor Xa (regardless of the length of the heparin molecule). Since heparin exists as a polymer with variable sizes (sugars), there are different forms that are utilized clinically and lead to different therapeutics effects.
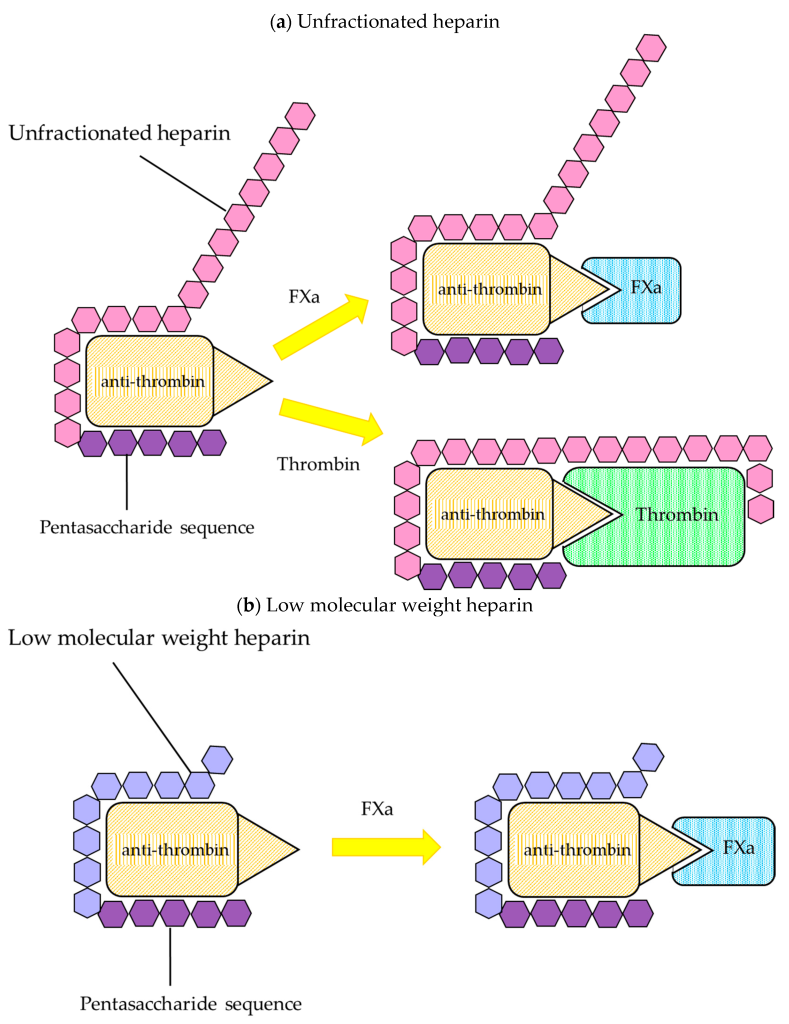
2.2.2.1 Unfractionated Heparins
Unfractionated heparin (UFH) is heparin that is isolated from porcine blood, and contains a complete mixture of sugars (the molecular weight of each heparin molecule ranges from 5000 – 15000 Da). This heparin is easier to generate since there are fewer purification steps. Furthermore, since this mixture contains both large and short heparin molecules, both thrombin and factor Xa are inhibited effectively. However, the variability in size of the molecules translates into variability in pharmacokinetic profiles following administration.
2.2.2.2 Low Molecular Weight Heparins
To compensate for the variability in pharmacokinetic profiles, the heparin can be further purified into a more homogeneous mixture. Low molecular weight heparin (LMWH) is generated such that the molecules have molecular weights lower than 8000 Da, which greatly improves the pharmacokinetic profile reproducibility/predictability. However, the larger portion of shorter heparin molecules also results in a reduced binding/inhibition of factor thrombin (since a smaller subset of the heparin molecules can carry out the bridging function). As such, LMWH is generally less efficacious than UFH.
2.2.2.3 Fondaparinux
Continuing with the theme of truncating the heparin molecule for more reproducible pharmacokinetic profiles, heparin can be further abridged to the pentasaccharide sequence. This version does not exist naturally and is only generated synthetically and is marketed as Fondaparinux. This is an extremely potent inhibitor, although it can only block the activity of factor Xa (and not thrombin).
2.2.2.4 Heparin Induced Thrombocytopenia
The purpose of heparin is to “thin” the blood and act as an anti-coagulant. However, there are scenarios (~5% of patients) when administration of heparin leads to the opposite effect. This is referred to as heparin induced thrombocytopenia (HIT) and there are two sub-types. Type I HIT is generally milder and occurs when heparin engages with a positively charged protein called platelet factor 4 (PF4) to create an IgG-antigenic complex. Type II HIT is usually an immune-mediated response as a result of the patient generating antibodies due to a component of the heparin injection (especially relevant since UFH or LMWH originate from another species). This can be life-threatening since the patient can enter a hypercoagulable state. In this case, usually the patient is treated with a substance that can neutralize the effects of heparin (which is a negatively charged species). In this case, protamine sulfate (a cationic peptide) is administered. It is important to note, that there are no cases of HIT observed with administration of Fondaparinux, but there are also no counteragents in case there is an overdose.
Challenges with the common anti-coagulants such as warfarin (which requires a high degree of personalization, monitoring, and has a slow onset) and heparin (which requires injections and has the potential of HIT) led to development of new inhibitors that could engage with thrombin or factor Xa. The goal for these drugs was to find a balance between efficacy, ease of administration, and onset of action. These drugs were developed as novel oral anti-coagulants (NOACs), although this term has fallen out of favour in place of DOACs (direct oral anti-coagulants).
2.2.3 DOACs: Direct Thrombin Inhibitors
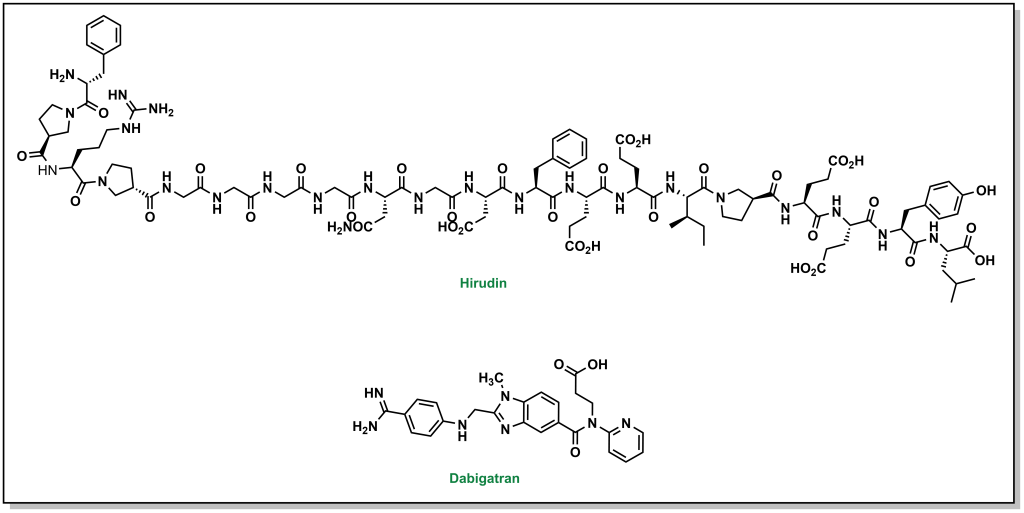
Thrombin is the next upstream target in the coagulation cascade, and the protein structure was known to have key distinct sites including an active site, exosite site 1 (fibrin binding site), and exosite 2 (heparin binding site). The first inhibitors were identified as derivatives of hirudin analogues (compounds isolated from the salivary glands of the leech Hirudo medicinalis) which correspond to ~20 residue synthetic peptides. These molecules directly bind to thrombin in an L-shape such that they blocked both the active site and exosite 1. Although these inhibitors were potent, as large peptides these molecules had to be administered subcutaneously. Newer generations of molecules are active as oral agents, such as dabigatran, which engages only at the active site of thrombin and contains a critical benzamidine group.
2.2.4 DOACs: Direct Xa Inhibitors
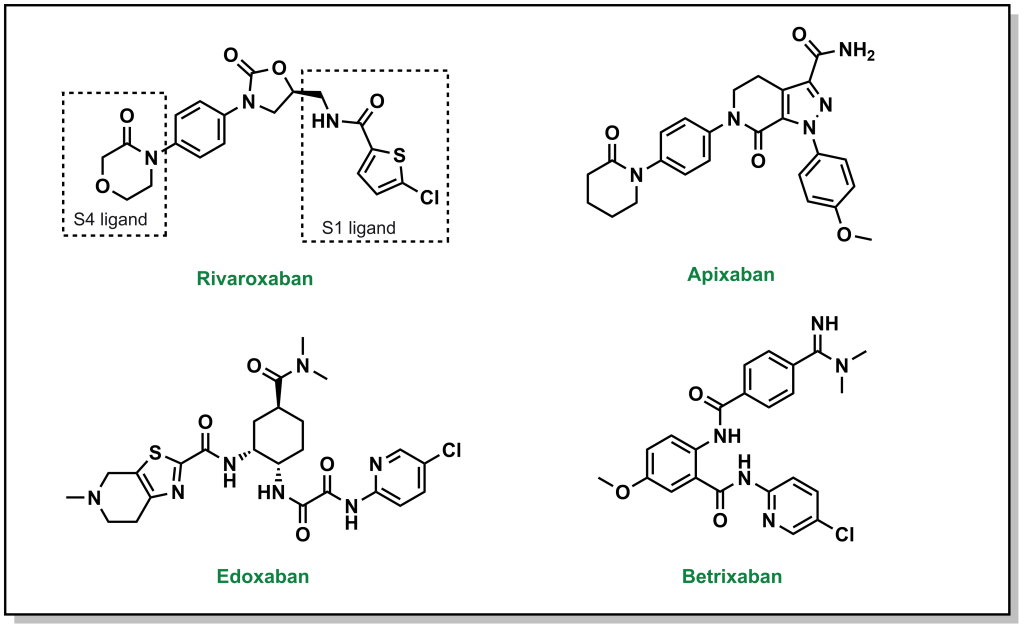
There are also DOACs that target factor Xa, and can be employed as substitutes for warfarin that have a reduced emphasis on blood clot testing, more predictable PK profiles, and a rapid onset of action. Similar to direct thrombin inhibitors, these molecules also bind in an L-shape across 4 sub-pockets of factor Xa (named S1, S2, S3, and S4). From a nomenclature viewpoint, these drugs are usually easily recognisable since they all contain ‘Xa’ in their name, such as rivaroxaban, aprixaban, edoxaban, and betrixaban. (Figure 2.6) The pharmacophore for these inhibitors is less readily identifiable, and is based on facilitating pi-pi stacking interactions between a phenyl ring of the inhibitors and the indole of Trp214 on factor Xa, as well as additional hydrogen bonding interactions throughout the molecule.
2.3 Summary
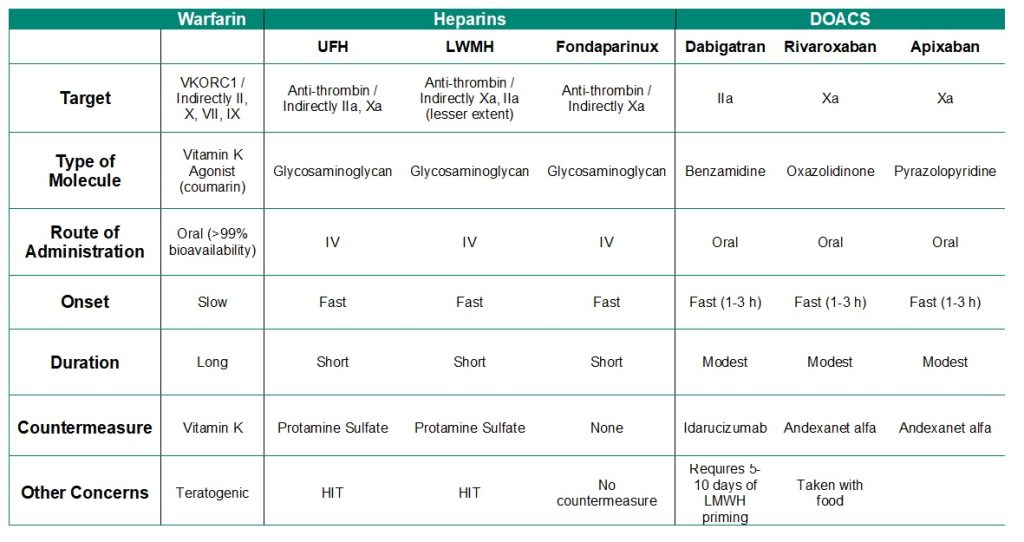
The following table summarizes important properties for blood anti-coagulant drugs.
Table 2.1: Summary of anti-coagulation agents.