6 Drug Modifications to Improve Stability
“You can’t use up creativity. The more you use, the more you have.”
– Maya Angelou
6.1 Isosteres and Bioisosteres
Improving ADME properties is a fundamental aspect of medicinal chemistry to limit the effects of first-pass metabolism while maximizing the efficacy of a drug candidate for its target. The general strategy to accomplish this is to employ isosteres or bioisoteres in a lead or drug candidate. While isomers refer to molecules that have the same number and type of atoms (albeit in different three-dimensional arrangements), isosteres refer to molecules that have different atoms but similar electronic properties. Likewise, bioisosteres have different atoms (which can lead to different chemical or physical properties) but share similar electronics or geometry, which results in the capacity of offering a similar biological function as the original ligand.
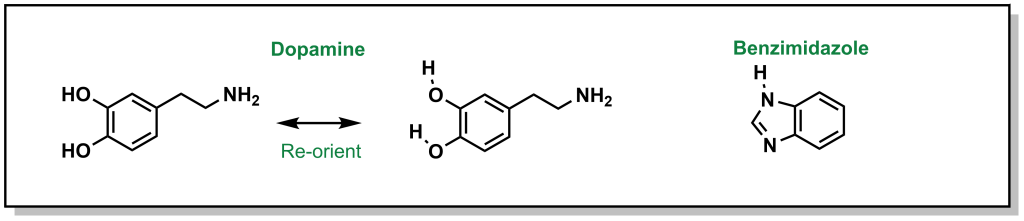
Throughout hit-to-lead and lead optimization, identifying bioisosteres that have functional mimicry is a key aspect of designing an SAR study. For example, replacing a thiol with a hydroxyl group preserves the use of an electronegative atom from the same group of the periodic table while offering an HBD. Some bioisosteres maybe non-obvious, for example, a catechol can be mimicked by a benzimidazole. This is because the neighbouring hydroxyl groups of the bis-phenol can participate in hydrogen bonding, which leads to a pseudo-five membered ring with a free hydrogen. In this way, the benzimidazole geometrically mimics this structure and contains a free hydrogen bond donor (Figure 6.1).
Over the past century, exploration of chemical space across hundreds of drugs has led to a ‘dictionary’ of bioisosteres that can be employed and evaluated in drug design. There are generally two types: classical and non-classical bioisosteres. Classical bioisosteres are structural analogs and are usually readily identifiable because they have similar sizes. Classical bioisosteres can be catalogued as monovalent/univalent (eg. CH3 ~ NH2 ~ OH ~ F ~ Cl), bivalent (eg. vinyl ~ carbonyl ~ imine ~ thioketone), trivalent (eg. alkynyl ~ nitrile), tetrasubstituted (tetrasubstituted carbon ~ tetrasubstituted nitrogen) and ring equivalent (eg. benzene ~ pyridine ~ thiophene). Non-classical analogues aim to mimic either the geometry or electronics/H-bonding capacity, but may look structurally distinct from the functional group being imitated. (eg. carboxylic acid ~ tetrazole). In either case, bioisosteres represent a strategy to preserve specific target-molecule interactions while modulating lipophilicity, solubility, acidity/basicity, and other properties.
6.2 Bioisosteres of Hydrogen
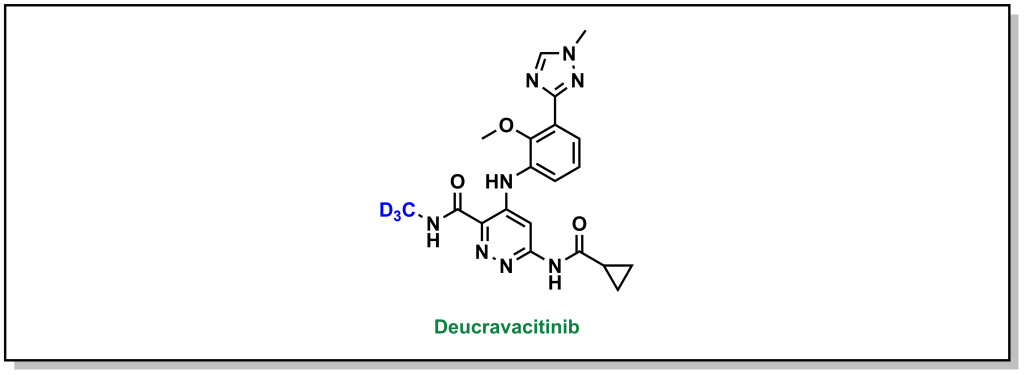
Depending on their location within a molecule, hydrogen atoms can sometimes be a metabolic soft spot. There are two main bioisosteres for hydrogen atoms. The most similar isostere is deuterium, a heavy hydrogen isotope (and is also non-radioactive and naturally occurring). As an isotope, the chemical properties of hydrogen are essentially preserved in replacement with deuterium. However, the additional neutron doubles the atomic mass which drastically alters the kinetics of a reaction (deuterium kinetic isotope effect). This becomes important if the hydrogen is involved in a catalytic step or as a hydrogen bond donor. Although they are naturally occurring, deuterium isotopes have a significantly increased costs associated with synthesis, and are not frequently employed as bioisosteres. Deucravacitinib is a selective Janus kinase (JAK) inhibitor of TYK2 that contains a magic-methyl moiety that is critical for activity (Figure 6.2). This methyl (located on the amide) is deuterated which blocks metabolism of the sp3 site (the C-D bond is ~5-10-fold stronger than the C-H bond due to lower vibration frequencies). Deuterium is the only isotope commonly considered in medicinal chemistry because the mass change is not as significant for larger atoms to substantially alter reaction kinetics (although other atomic isotopes may be employed in drugs for purposes such as imaging or radiation therapy).
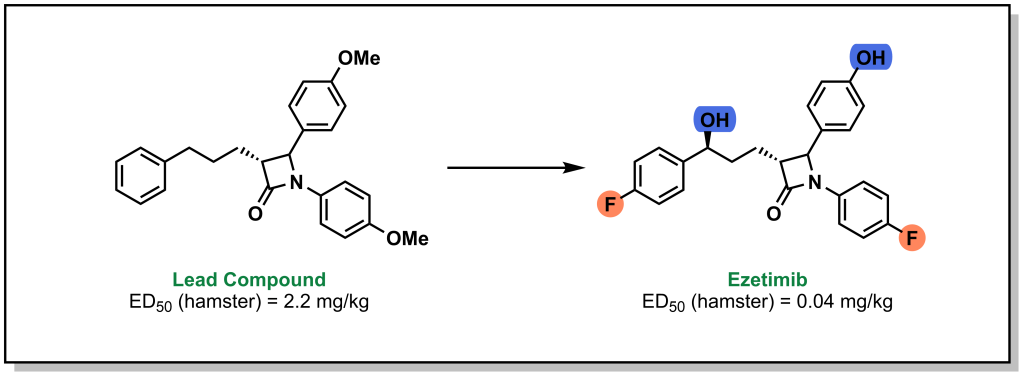
A more commonly employed isostere for hydrogen is fluorine. This may seem unusual as fluorine is a highly electronegative halogen. However, the electronegativity of fluorine reduces the size of the electron cloud leading to similar sizes (carbon bond lengths of 1.20 Å [C-H] vs 1.35 Å [C-F]). The electronegativity difference makes the carbon-fluorine bond the strongest bond in organic chemistry because of the partial ionic character of the bond. This makes C-F groups highly resistant to oxidation by CYP enzymes. For example, the cholesterol absorption inhibitor, ezetimibe, was generated with fluorine atoms at the para-positions on both free benzene rings to reduce the metabolic instability of the para-hydrogen atoms (Figure 6.3). Importantly, the electron-withdrawing inductive effects of fluorine should also be considered when substituting a fluorine into a molecule (for example the impact on a nearby HBA).
6.3 Bioisosteres of Phenyl Rings
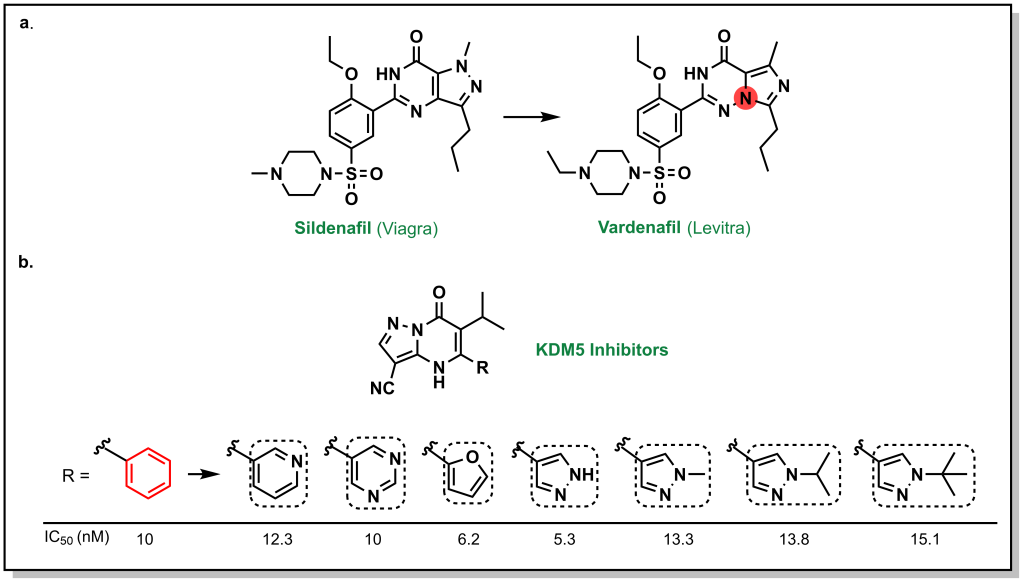
Phenyl rings are extremely common in drug discovery and possess a planar structure and engage in multiple hydrophobic interactions. Classical bioisosteres of phenyl rings aim to preserve the aromaticity and/or planar structure such as the previously described example of Viagra to Levitra involved a straightforward substitution of a carbon atom for a nitrogen atom (Figure 6.4a). Other examples include the SAR study performed by Liang et al (2016) and summarized by Subbaiah et al (2021), these hetero-atom substitutions on a phenyl ring in KDM5 inhibitors can facilitate new interactions and provide a scaffold for additional substitutions (Figure 6.4b).
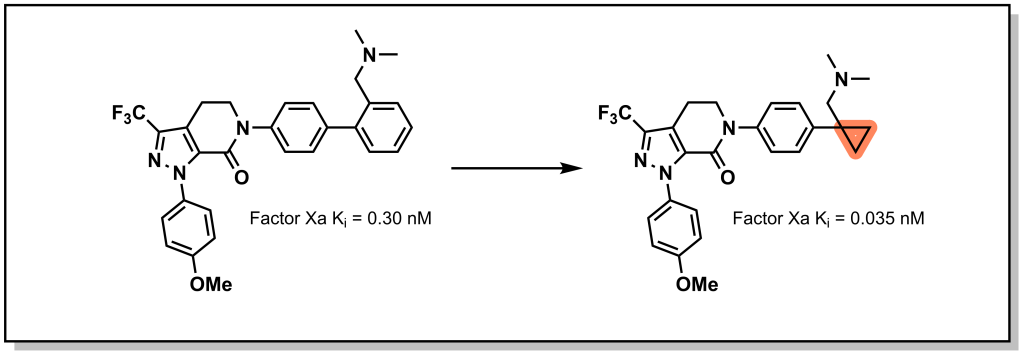
There are less obvious bioisosteres for phenyl rings where the goal is to preserve the exit vector of a substituent. For example, cyclopropane can be employed as a bioisostere in some cases. In Factor Xa inhibitors generated by Brystol Myers Squibb, replacement of the phenyl ring with a cyclopropyl ring maintained the orientation of the tertiary amine, but substantially reduced the molecular weight thus increasing ligand efficiency (Figure 6.5). In this case, the phenyl ring was not functionalized, and the efficacy of these substitutions depends on the chemical species that can be accommodated in the pocket as well as the resulting changes to LogP / lipophilicity.
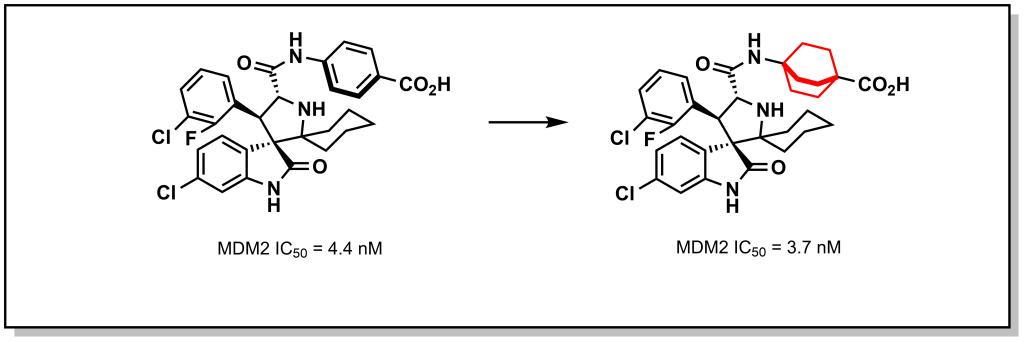
Other non-classical bioisosteres of phenyl rings that have emerged include saturated bi- and polycyclic-rings such as cubanes, bi-cyclopentanes, and bi-cyclohexanes. These replacements are known to increase both metabolic stability (reduced 1,4-quinone formation) and water solubility (reduced π-π stacking interactions) while preserving dihedral angles (180°). These polycyclic rings show optimal functional mimicry when the original role of the phenyl ring is as a spacer (as opposed to participating in key interactions such as aryl-protein π-π stacking). For example, replacement of the phenyl ring with a bicyclohexyl group in the MDM2 inhibitor was shown to maintain potency but with greatly increased metabolic stability leading to a 6-fold increase in PK exposure (Figure 6.6). For these substitutions it is important to consider the linker length in deciding which bi-cyclo ring to install. For example, the end-to-end distance of C1-C4 atoms of benzene is 2.82 Å, while the comparable distance in a bi-cyclohexane, bi-cyclopentane, and bi-cyclobutane is 2.72 Å, 1.70 Å, and 2.60 Å respectively. Incorporation of these (and other) polycyclic rings can potentially enable precise adjustment of linker lengths.
6.4 Bioisosteres of Carboxylic Acids
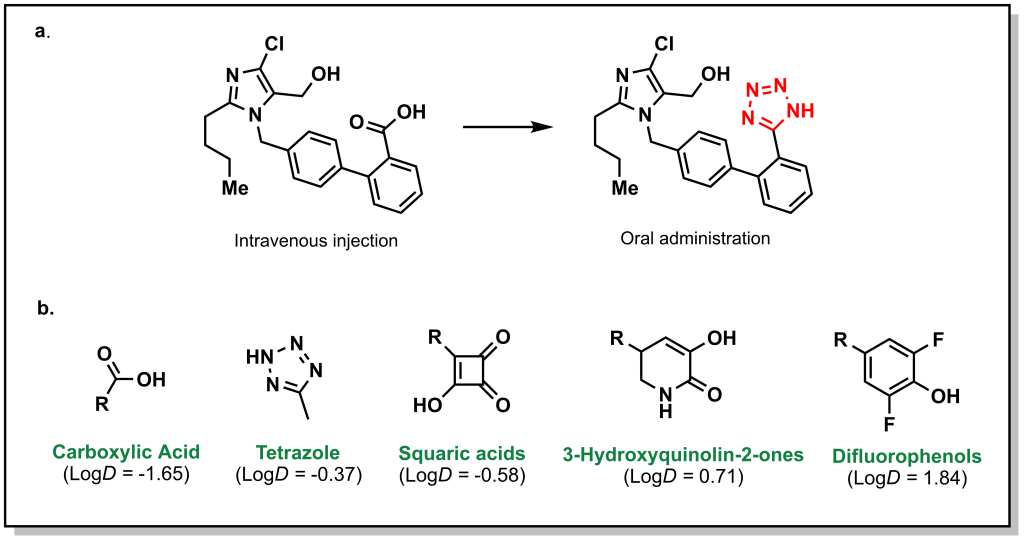
Bioisosteres for carboxylic acids have been heavily explored because of the anionic nature of the functional group which exhibits large (and usually unfavourable) effects on permeability and PK. The negative charge of the carboxylate group is delocalized over two oxygen atoms and responsible for potency with many protein targets via ionic interactions. Therefore, bioisosteres usually require an acidic proton as well as a two-point interaction profile to function as a suitable surrogate. The most common (non-classical) bio-isostere is a tetrazole, which contains an acidic proton (pKa ~ 4.5) but is ~10-fold more lipophilic. For example, potent inhibitors of AT1 (GPCR) receptor were developed based on a carboxylic acid-receptor interaction but only available by IV injection (Figure 6.7a). Replacement with a tetrazole improved binding but also enabled oral administration due to increased lipophilicity and led to the antihypertensive drug, losartan. There are hundreds of different types of carboxylic acid bioisosteres that have been explored, such as difluorophenols, squaric acids, and hydroxyquinoline-2-ones (Figure 6.7b).
6,5 Bioisosteres of Amides
Amide bonds may be metabolically unstable in drugs due to their enzymatic sensitivity towards proteases (often targeted towards peptide bonds). Bioisosteres of amides are designed to mimic the polarity as well as the planar geometry of the amide bond. Functional groups such as ketones, carbamates, ureas, and esters, can all mimic the polarity of the amide bond as well as the geometry. Triazoles can mimic both the H-bond acceptor and H-bond donor properties while maintaining similar spacer distances. In a similar manner, oxazoles, imidazoles, pyrazoles, thizaoles, can also substitute as cyclic replacements for an amide. Therefore, there are multiple functionalities that can serve as appropriate substituents, which provide opportunities for custom tailoring.
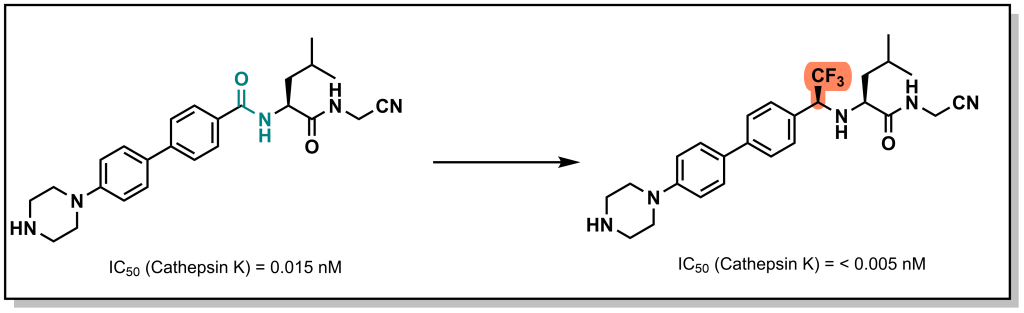
Less conservative changes have more direct atom substitutions but attempt to preserve the geometry and HBD and HBA character. For example, trifluorethylamine can be used to replace the amide bond in the Cathepsin K inhibitors developed by Merck (Figure 6.8). This allows for reducing the basicity of the amine while still enabling HBD properties and preserving the 120° bond geometry and polarity (the C-CF3 bond has been determined to be as polar as the carbonyl C=O bond).
Oxetanes are also potential replacements for the carbonyl of an amide since they are stable and are significantly polar. The positioning of the oxygen within a 4-membered ring results in a larger space for the lone-oxygen pairs, potentially improving their role as HBAs. Oxetanes can also be used as bioisosteres for gem-dimethyl groups where they adopt similar steric volumes without the lipophilicity increases of the methyl substituents. These functional groups also have increased sp3 character with reduced susceptibility to CYP oxidation.
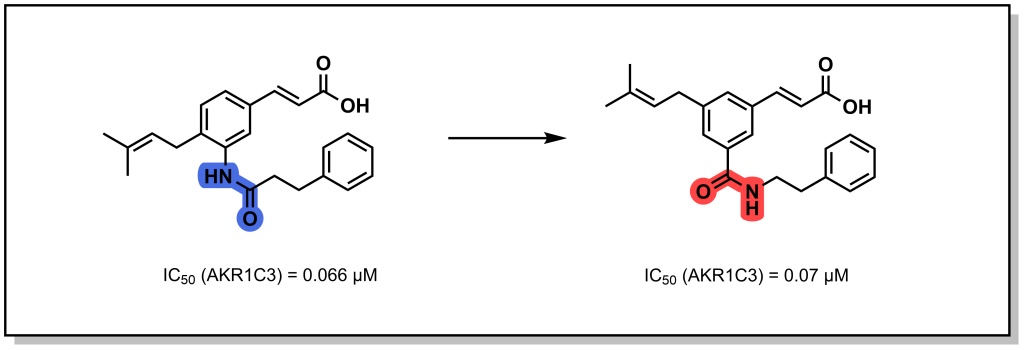
An alternative strategy involves transposing the amide, which increases resistance to protease activity (metabolic stability) and can have limited effects on target engagement, H-bonding, and conformational mobility. For example, transposition of the amide in the bacterial AKR1C3 inhibitor substantially improved compound selectivity through improved HBD contacts.
6.6 Overall Approach to Bio-isosterism
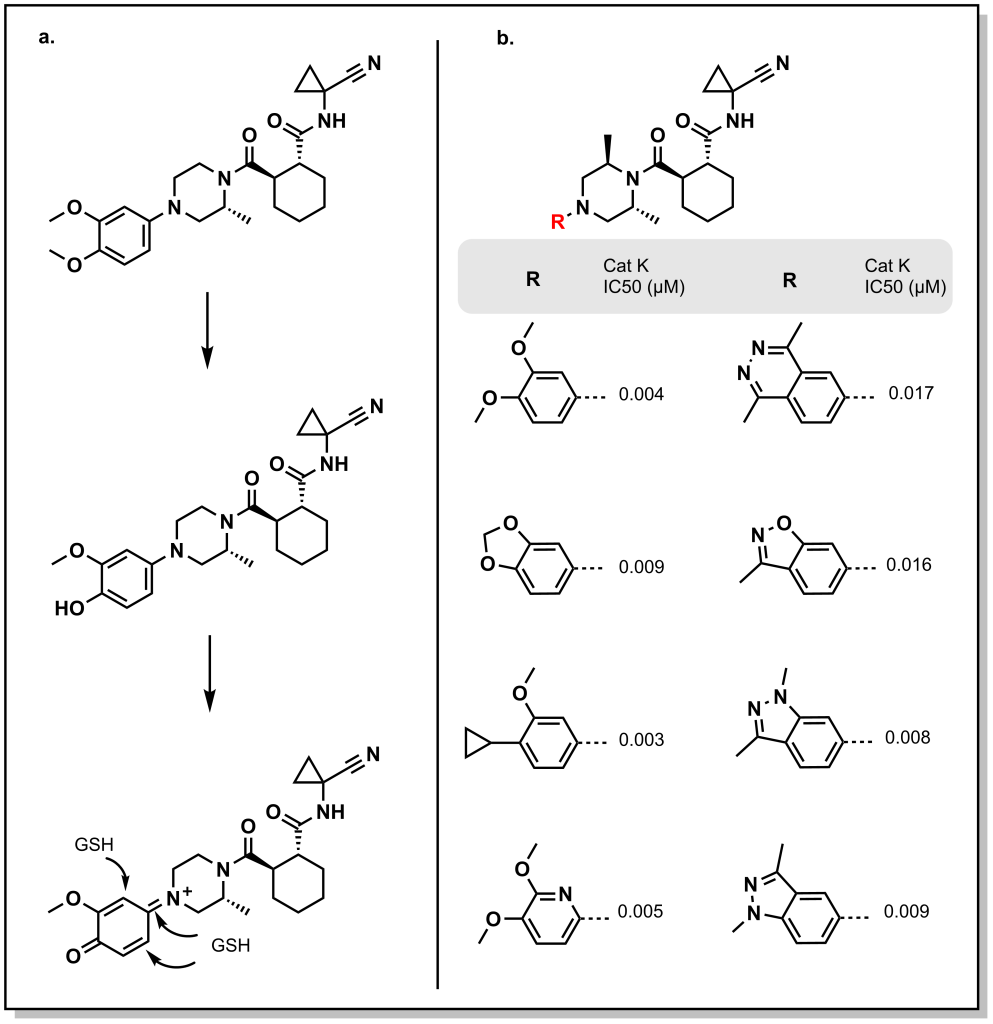
In general, the main goal for bio-isosterism involves recognizing metabolic or permeability soft-spots in the molecule and identifying appropriate functional groups that can serve as a replacement. This can involve more sophisticated approaches that are not simply a one-for-one substitution. For example, Astra Zeneca was generating cathepsin K inhibitors for treatment of osteoporosis and other bone related indications. Their lead compound, balicatib, was shown to be potent, leveraging interactions of the cyclohexyl bisamide moiety, although it had poor PK properties and was susceptible to glutathionation. (Figure 6.10a) Structural data suggested that modifications to the phenylpiperazine rings would not disrupt the core pharmacophore. Several bioisosteres were attempted to mimic the capped-bisphenol, which was found to be susceptible to oxidation and glutathione reactivity. For example, tying back the O-methoxy groups into a 5-ring structure resulted in additional Phase I metabolism. This was similar to the cyclopropyl group which is a comparable isostere for an oxygen atom (in size). Installing 5- or 6- membered rings with heteroatoms creates unique positions for the lone-pairs and electron density, with the indazole demonstrating the best overall properties. (Figure 6.10b)
6. 7 Structural Alerts
There are literally hundreds of bioisosteres that can be employed in drug discovery and being able to recognize and suggest functional groups is a key part of the design process. Equally, identifying functional groups that can lead to toxicity or reactivity is a critical aspect. These functional groups are referred to as structural alerts on a molecule and can indicate potential mutagenicity, hepatic toxicity, renal toxicity, phospholipidosis, etc. Importantly, these functional groups can be structural alerts for toxicity in one drug, but not another, because of competing factors such as sterics or electronics that may block metabolic bioactivation.
6.7.1 Aromatic N-H and O-H
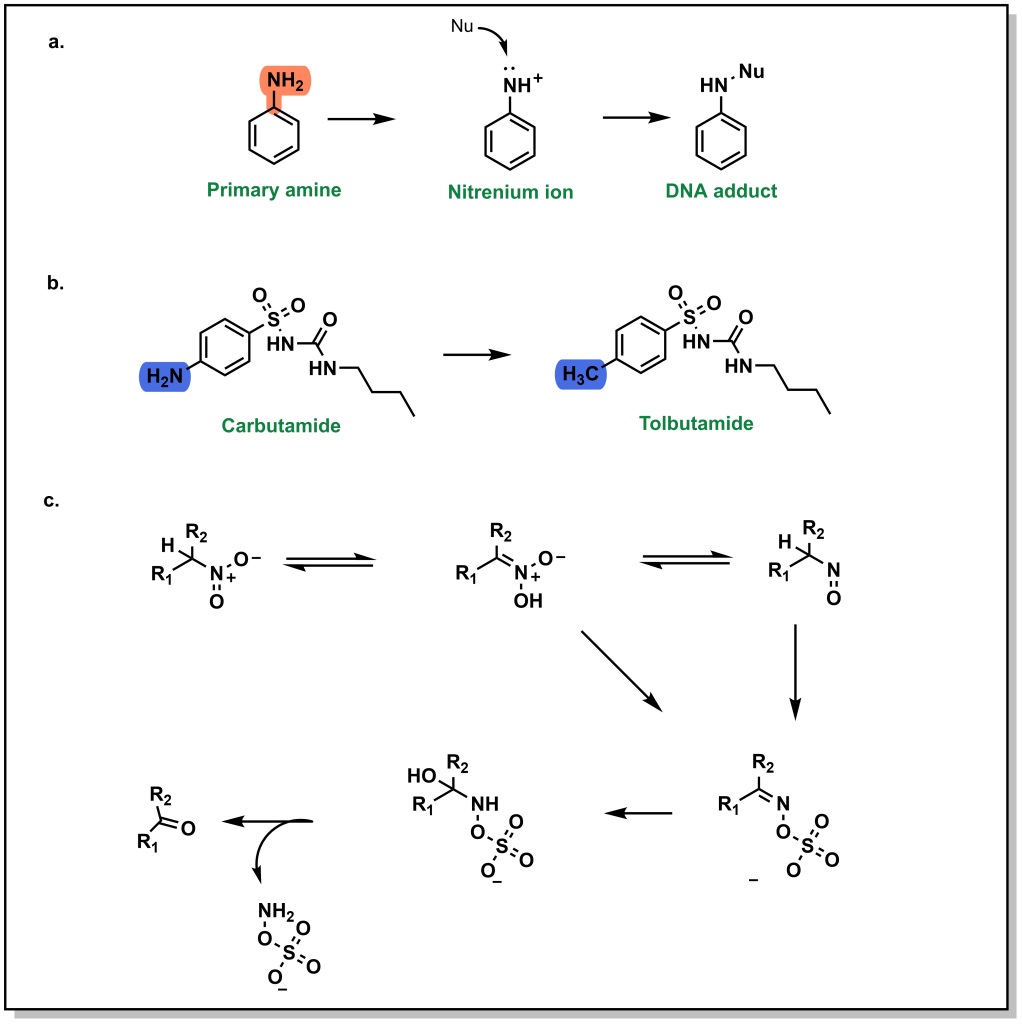
Aromatic N-H groups may be susceptible to nucleophilic attack, which generates a reactive nitrenium ion that can react with important biomolecules such as DNA resulting in mutagenic toxicities. (Figure 6.11a) Replacement of the aromatic amine can remove the structural alert such as substituting a methyl group on carbutamide to give tolbutamide (sulfonylureas for hyperglycemia). (Figure 6.11b) Non-aromatic amines can also be oxidized to form nitroso groups which are toxic metabolites (and therefore structural alerts). Similarly, nitro- groups are poor HBA and readily oxidized to nitroso-groups and often avoided in drug discovery. (Figure 6.11c)
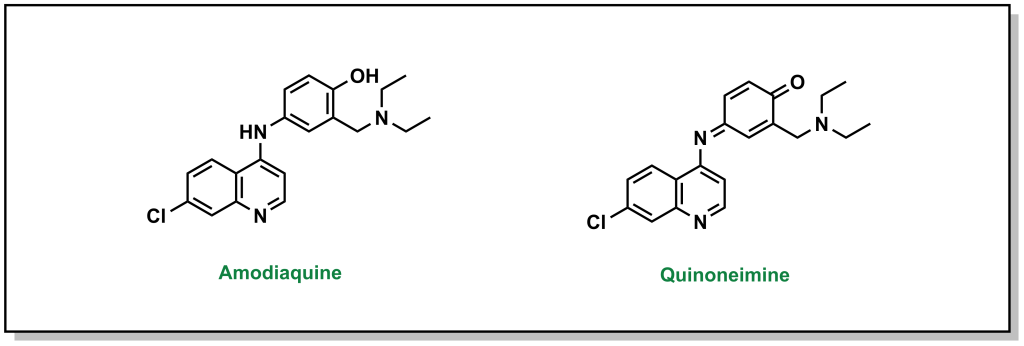
Similar to primary aromatic amines, aromatic alcohols are also structural alerts due to the formation of quinones, which are 1,4 Michael acceptors. These species are highly electrophilic and can react with nucleophilic biomolecules, leading to covalent attachment and irreversible damage. (Figure 6.12)
6.7.2 Unsubstituted Rings
Structural alerts can also apply to aromatic and aliphatic ring systems, where unsubstituted rings are often a key indicator of potential metabolism. Cyclohexyl groups are often oxidized at the 4-position initially, and a common protection strategy is to block this position with a bioisostere such as fluorine. Similarly, changing a piperidine ring for a morpholine ring can offer solubility advantages while blocking the metabolic soft spot. Additional changes can also be feasible such as introducing ring strain or sp2 character. Likewise, unsubstituted benzene rings are structural alerts as these are subject to oxidation via epoxidation. The most common strategy is to deactivate the ring with an electron-withdrawing group, particularly at the para- position. Introduction of hetero-atoms in the ring can also be helpful, and the more nitrogen atoms present in the ring, the more resistant they are to CYP-mediated oxidation.
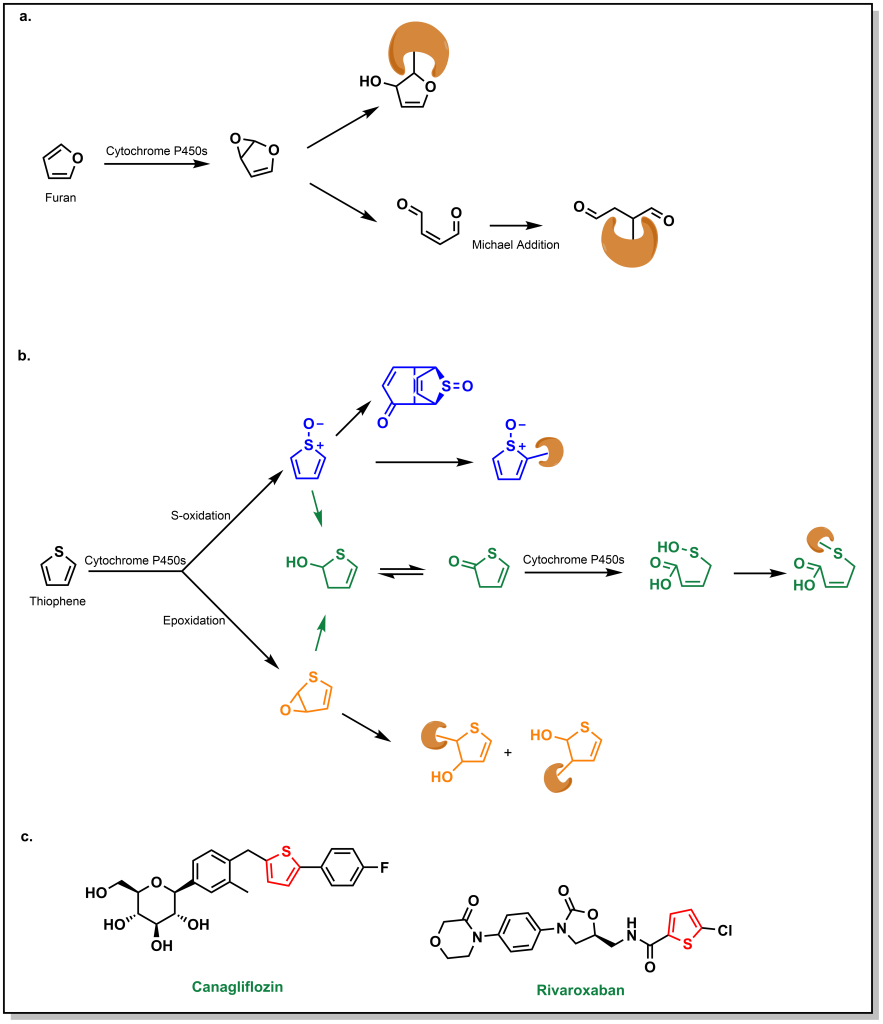
6.7.3 Furans, Thiophenes, and Thiazoles
Five-membered hetero-substituted rings can also be structural alerts and can lead to protein or biomolecule covalent modification. (Figure 6.14a,b) Furans are substrates for oxidation (similar to benzene rings), which can lead to hydrolysis and generation of highly electrophilic species. Thiophenes can also lead to epoxidation but result in free thiols that lead to hepatotoxicity. Importantly, some of these structural alerts are dormant in different molecules. For example, the thiophene moiety is dormant in both canagliflozin (a sodium glucose co-transporter inhibitor) and rivaroxaban (a Factor Xa inhibitor). For canagliflozin, metabolism predominantly occurs via UGT enzymes on the glucose moiety. For the rivaroxaban, metabolism occurs on the morpholinone ring system. Both examples indicate that if there is steric hindrance or other sites that are more susceptible to oxidation, this can bypass potential structural alerts on a molecule. (Figure 6.14c)
6.8 PAINS Molecules
In addition to recognizing structural alerts to toxicity, there are additional classes of Pan-Assay INterference compounds which are referred to as PAINs alerts. These are functional groups that often yield false positive hits in assays. PAINs molecules are important to recognize early-on in the hit identification process, because if they are advanced further in the drug discovery pipeline, significant resources can be invested into a non-binding compound. Furthermore, since these molecules may exhibit non-specific binding to the target, they cannot be optimized to improve binding. For instance, a quinone is highly reactive and can react with a protein non-specifically, disrupting its secondary structure. In an activity assay, this loss of secondary structure could be observed as a loss of activity, which would be similar to what would be observed if a molecule bound to the protein as a true hit. PAINs molecules become particularly important when a high-throughput screens of thousands of compounds are performed.
There are several recognized molecular scaffolds for PAINs activity. However, sometimes a molecule exhibits PAINs activity in a screen against a specific target but is an active-binding participant in the pharmacophore of another molecule. For example, if the scaffold has metal-chelating potential (such as a free-thiol), it could show PAINs activity against an array of metalloproteins but not against non-metalloproteins. Therefore, every drug program needs to evaluate PAINs moieties independently and early-on in the process. Orthogonal binding and activity assays are important to confirm hits, before embarking on a full SAR campaign.
