4 Drug Absorption and Distribution
“Science is magic that works.”
– Kurt Vonnegut
4.1 Introduction to Pharmacokinetics and Pharmacodynamics
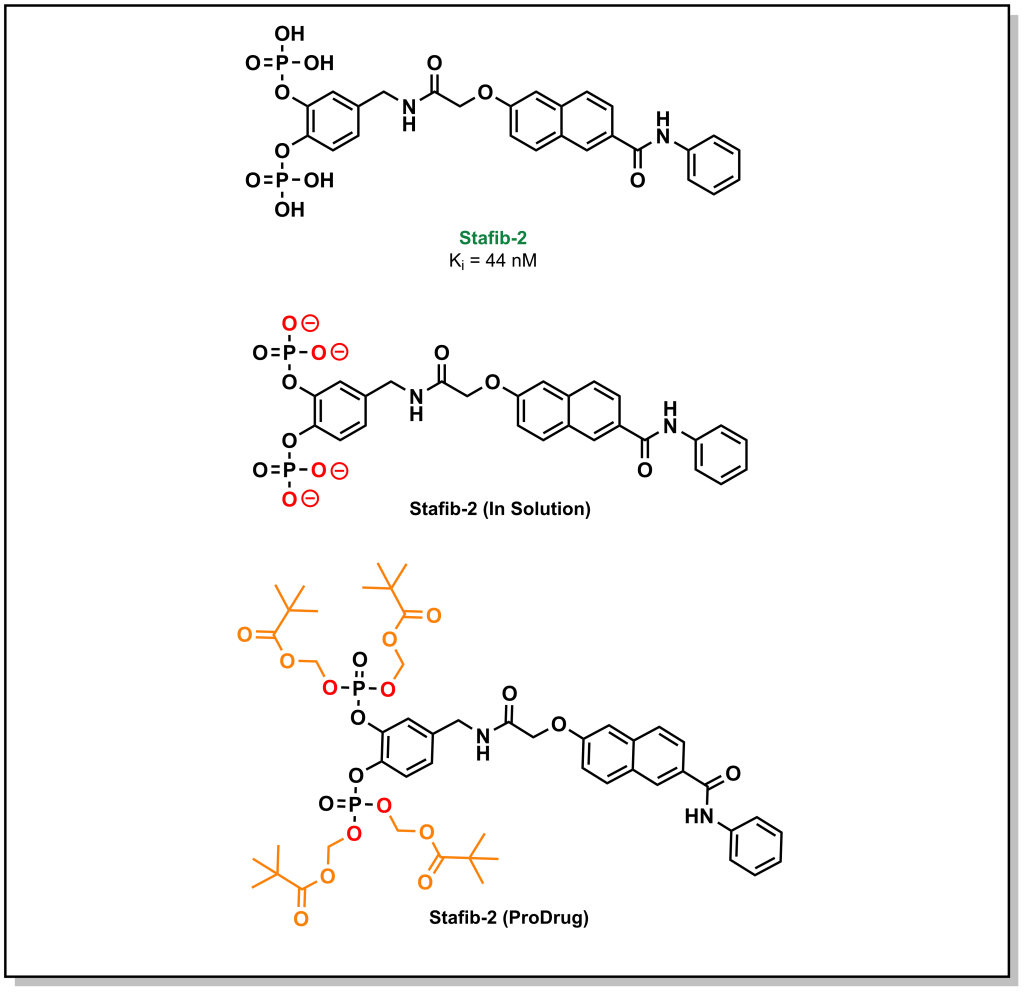
A large emphasis of the early stages in drug discovery is placed on obtaining a lead molecule that can elicit a potent effect on a target of interest. Once a lead is generated, the next stage involves exploring the pharmacological utility of the molecule. For instance, a lead may exhibit high potency and selective target engagement in vitro but is not capable of exerting a biological effect in cellulo or in vivo. The molecule, staffib-2, provides a representative example of this conundrum. Stafib-2 was designed to target the SH2 domain of STAT5B, a transcription factor upregulated in multiple cancers. The origins of the molecule emerged from natural products and catechol bisphosphate derivatives, which can mimic phospho-tyrosine moieties (SH2 binding scaffolds). The structure is chemically optimized to exploit a π-π stacking interaction of the catechol with a tryptophan in the SH2 domain, along with hydrophobic interactions of the transposed benzamide with a nearby phenylalanine (Figure 4.1). The resulting molecule demonstrated potent inhibitory activity (Ki = 44 nM) with >50-fold selectivity over the closely related STAT5A isoform. Based on these data, this is an excellent hit molecule, but it demonstrates extremely poor activity in cellular assays. This lack of activity arises from the pKa values of the phosphate groups (1.5 and 6.3), which are indicative of the ionization state of the molecule at physiological pH. The di-anionic charge on each phosphate group prevents the molecule from effectively crossing the phospho-lipid bilayer, and these properties had to be masked in the form of a pro-drug in order to obtain phenotypic efficacy.
Therefore, it is not always sufficient to identify a molecule with excellent potency and selectivity. The effects that physiological processes of the body can have on the drug, as well as the effects that the drug can have on the body need to be considered. These effects are called pharmacokinetics (PK) and pharmacodynamics (PD), respectively. Understanding and optimizing both the PK and PD properties of a molecule forms a large component of medicinal chemistry and the drug discovery pipeline.
4.2 DMPK & ADME
Drug Metabolism and Pharmacokinetics (DMPK) refers to the study of how the body will engage and interact with a drug. PK is extremely important in drug discovery, as a molecule that enters the body will be treated as a foreign entity. Hence, it will need to survive the multiple defense mechanisms that the human body has evolved to deal with xenobiotics, in order to reach its specific target and achieve therapeutic effects. Pharmacokinetics is often sub-divided into different processes which include drug Absorption, Distribution, Metabolism, and Elimination, and are collectively referred to by the acronym ADME. Although ADME is often presented and treated as distinct phases of the overall DMPK process, these phases are all interrelated. The focus for medicinal chemistry will be towards absorption and metabolism whereas distribution and excretion are largely biology/physiology-based topics.
4.3 Absorption In Cellulo
Initial exposure to a drug and its uptake into the body is called absorption. There are different considerations for drug absorption that depend on the complexity of the biological and molecular system under evaluation.
4.3.1 Cellular Membranes
For an in cellulo experiment, the molecule is administered directly to the local environment of the cell. If the target exists on the outer layer of the cell surface (e.g. a GPCR), the molecule does not need to transverse the cell membrane and can effectively bypass a number of permeability challenges. However, if the target exists within the cell, a drug will need to cross the membrane to reach its intracellular target. Recall that the structure of a cell membrane is formed by a phospholipid bilayer intercalated with proteins. As such, the negatively charged phospho-lipid headgroups are facing both the extracellular and intracellular matrix and create a highly non-polar “sandwich” that segregates the cell from exterior contents. The biochemical implications of this bilayer structure impose unique property requirements for drugs/compounds traversing the membrane.
4.3.2 Transport – Passive Diffusion
Passive diffusion refers to the spontaneous movement of a substance from a region of high concentration to a region of low concentration. For a drug that is administered, this means that (at least initially) there is a high concentration outside the cell and a low concentration inside the cell. Based on concentration gradients alone, a drug would therefore be energetically favoured to enter the cell. However, polar or charged molecules cannot readily partition into the hydrophobic lipids of the bilayer and diffuse at a substantially lower rate. This can be quantitively described by Fick’s law of diffusion.
As a general trend, neutral compounds will have a higher rate of passive diffusion, followed by bases, zwitterions, and acids. Passive diffusion is the predominant mechanism of transport for lipophilic compounds. Although bases carry an ionic charge, they are not as energetically disfavoured for passive diffusion since the positive charge of the molecule can associate with the negatively charged phospho-lipid headgroups to initiate the diffusion process. Similarly, zwitterions (positively and negatively charged species) can also associate in this manner and have a net neutral charge. Acids (negatively charged species) are especially challenging to transverse the membrane via passive diffusion due to repulsive electrostatic interactions with the phospholipid headgroup. However, this impermeability of anionic species to the lipid bilayer can be an effective tool in drug discovery. Consider the example of Stafib-2 above. The negatively charged phosphate groups prevent the uptake of the compounds into the cell interior. These charges can be masked by generating phosphate esters. This creates a neutral species that is now capable of crossing the membrane via passive diffusion. However, Stafib-2 needs to be negatively charged to engage with the positively charged sub-pocket of the STAT5B SH2 domain. Esterases (which are located predominantly within the intracellular matrix) can hydrolyze the compound into its active anionic form once it enters the cell. This negatively charged species can now engage with the target. Furthermore, the negatively charged species becomes trapped within the intracellular compartment and cannot passively diffuse out of the cell. Hydrolyzing the ester also reduces the concentration of drug-ester inside the cell continuing to pull the equilibrium towards compound influx. This strategy of generating an inactive species that is later converted to an active form, is a called a pro-drug approach.
4.3.3 Transport – Facilitated Diffusion
As depicted above, not all drugs (or organic compounds) are amenable to passive diffusion. Cells have developed multiple strategies for the transport of compounds that cannot be readily absorbed because they are too large or too polar. This includes a number of transporter proteins that can be hijacked by drugs for cellular import. These specialized transport mechanisms can generally be stratified into two streams, facilitated diffusion and active transport. Notably, both of these processes rely on a transporter, and therefore the protein can be saturated which can limit transport at high concentrations.
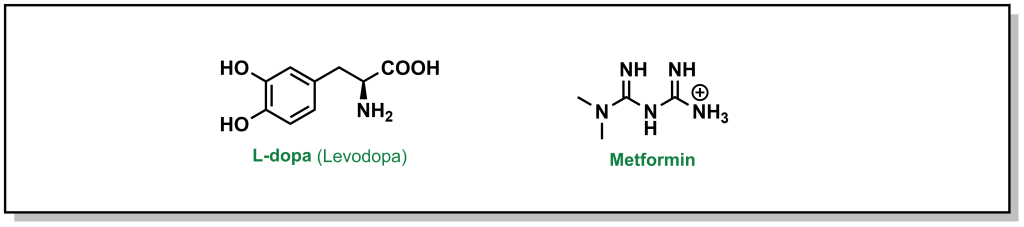
Facilitated diffusion is a passive process, where a molecule will move down a concentration gradient, but it requires a protein to mediate transport. As such, this is a selective process, as only specific chemotypes are permitted passage. Different drugs can leverage the use of known transporters. For example, L-dopa (Levodopa) is a drug used for the treatment of Parkinson’s disease that is too polar to passively diffuse across the blood brain barrier membrane (Figure 4.2). The amino acid structure of the drug can exploit the LAT1 transporter (L-type amino acid transporter) to enter the cell. However, utilizing natural transporters can also lead to challenges, as dietary consumption of protein sources (ie. amino acids), can compete with L-dopa and saturate the transporter, indicating patients have to monitor the timing of L-dopa dosing with their meals. Another example involves the most common blood glucose lowering agent for Type II diabetes, metformin (Figure 4.2). Metformin is a positively charged biguanide that cannot passively cross the membrane. Metformin enters cells via the Organic Cation Transporter-1 (OCT-1) which enables transport of positively charged species.
4.3.4 Transport – Active Transport
In contrast to passive or facilitated diffusion, active transport requires energy input to transport a molecule against its concentration gradient. The energy is provided in the form of ATP hydrolysis or an electrochemical/concentration gradient. In drug discovery, active transport is most relevant in terms of the efflux of xenobiotics (drugs) out of the cell. The Multi-Drug Resistance (MDR) proteins are a family notable for their ability to mediate the effluxing of drugs from cells, preventing their cellular accumulation, and augmenting disease cell survival.
Multi-drug resistance can be an especially challenging aspect in drug discovery and represents a general mechanism where the lifetime of a drug inside a cell is not extensive enough to carry out its therapeutic effects. It is important to note that this is different from specific types of drug resistance that can result from mutation of a protein to prevent action of the drug on its target (eg. the gatekeeper mutation in EGFRT790M discussed in Section 1, blocks the action of the kinase inhibitor, afatinib).
Although it is non-obvious whether a molecule is a substrate for an efflux-transporter, there are considerations to help limit drug-interactions with transporters. In general, efflux pumps effectively transport molecules with the following properties out of the cell:
- Lipophilicity
- Planar Structures
- Molecular Weight < 800 Da
- Weakly cationic
Unfortunately, these are also heavily desired properties for small molecule inhibitors. There are five protein families from the drug/metabolite transporter (DMT) superfamily.
- ATP-binding cassette (ABC) Transporters: This is one of the largest and most ancient protein superfamilies. The architecture of a conventional ABC transporter includes 2 transmembrane domains and 2 nucleotide binding domains that bind and hydrolyze ATP to facilitate conformational changes and pass substrates through the membrane. ABC transporters have diverse functions, and a subset of these proteins have drug efflux capacity. Multidrug resistance protein (ABCC1 or MRP1), and breast cancer resistance protein (ABCG2 or BCRP) confer resistance to several drugs. However, the most notable example is ABCB1, also called P-glycoprotein (P-gp) or MDR1, which poses a large obstacle in overcoming drug resistance for a broad array of structurally-diverse compounds (see below). P-gp is the most relevant drug efflux protein and specific assays evaluating P-gp activity are often incorporated into drug discovery pipelines.
- Major Facilitator Superfamily (MFS) or Uniporter-symporter-antiporter family: Similar to ABC transporters this is the second largest family of membrane transporters and is found in bacteria, archaea, and eukarya. Together with ABC transporters, these groups encompass more than half of all known membrane transporters. Unlike ABC transporters, these proteins rely on electrochemical gradients to facilitate transport and are usually single-polypeptides.
- Multiple Antimicrobial Extrusion Family (MATE) transporters: This is a class of secondary active transporters that are largely involved in transport of cationic compounds and are present in all domains of life. MATE transporters are often attributed to multi-drug resistant hospital infections due to their expression in Staphylococcus aureus. In humans, these transporters are mostly present at the brush-border of the kidneys. Similar to MFS transporters, typical MATE transporters contain 12 transmembrane helices.
- Resistance-nodulation-division (RND) Transporters: These transporters are found in (Gram-negative) bacteria as well as archaea and are particularly important in microbial xenobiotic defense/efflux. These pumps are asymmetric trimers that derive energy from proton gradients.
- Small Multidrug resistance (SMR Transporters): These are bacterial transporters comprised of four alpha helices that also derive energy from electrochemical proton gradients. These transporters focus on the movement of lipophilic compounds and quaternary ammonium compounds.
4.3.4.1 P-glycoprotein (P-gp)
P-glycoprotein is the most clinically relevant drug transporter due to its broad specificity and presence in the key tissues for ADME including the gastrointestinal tract, blood-brain-barrier, kidneys, and liver. P-gp acts in a unidirectional manner to extrude substrates outside of the cell, which can effectively topple a lead candidate in drug discovery pipelines. There are extensive published lists of drugs that are known to be actively effluxed by P-gp and 25% of all oral lipophilic drugs are known P-gp substrates.
P-glycoprotein affects the rate and concentration of drugs diffusing across the basolateral membrane of the intestine. For example, in an enterocyte (cells along the small intestinal lining), P-gp is expressed at the brush-border (site of absorption). This facilitates the removal of xenobiotics almost immediately and prevents their absorption. Another key site of P-gp expression is at the blood-brain-barrier (BBB), which represents a number of tightly packed cells that surround the central nervous system (CNS). These endothelial cells of the BBB contain P-gp that effluxes compounds and presents a major challenge in drugs accessing the CNS tissues. In drug-discovery pipelines, P-gp efflux is often evaluated through an MDCK1-MDR1 assay which involves Madin Darby Canine Kidney (MDCK1) cells transfected with the human MDR1 (P-gp) gene grown in a monolayer. An efflux ratio of < 2 indicates the compound is not a substrate for P-gp.
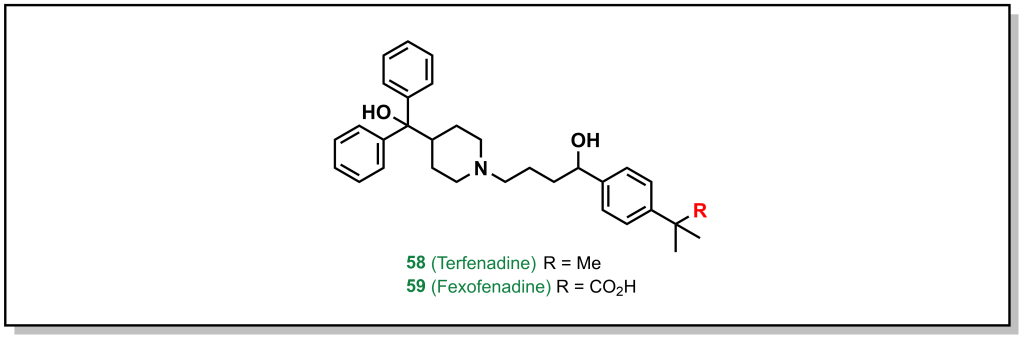
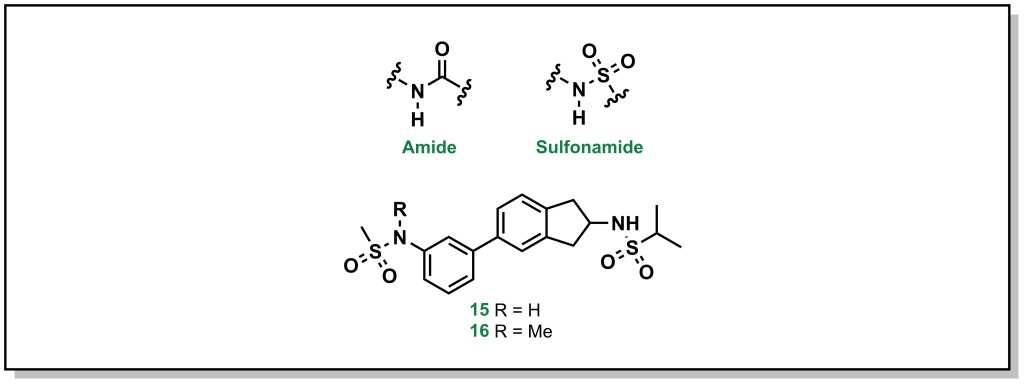
Amgen’s medicinal chemistry analysis of 4176 drugs demonstrated that 52% of P-gp substrates showed tPSA > 90 Å2 and 57% of P-gp substrates showed > 2 HBDs. Additionally, highly lipophilic drugs with LogP > 7 were P-gp substrates. These trends provide potential approaches to evade P-gp efflux, which are focussed on reducing HBD character as well as acidic/basic groups. Substrates that contain nitrogen atoms found in amines, amides, sulfonamides, and heterocycles, as well as oxygen atoms in hydroxy and carboxylic acids are also alerts for P-gp efflux. Although it is not a strict guarantee that molecules with these functional groups will be effluxed, if it is possible to remove the offending functional group without significant loss of activity, this is the best option. For example, removal of the carboxylic acid group from fexofenadine reduces P-gp efflux in terfenadine (anti-histamines for allergy medication; Efflux Ratio = 2.9 [Terfenadine] vs 6.8 [Fexofenadine], Figure 4.3). However, this is not always possible, in which case altering the hydrogen bond donor needs to be examined, such as: i) The proton can be directly removed (such as via alkylation) or ii) it can be masked. There are an array of different strategies to accomplish this for each functional group.
4.3.4.1.1 Amides and Sulfonamides
Amide and sulfonamide groups have a free N-H that is capable of functioning as a hydrogen bond donor. In specific substrates, it is straightforward to simply remove the proton by methylation, as with the AMPA receptor inhibitors developed by GSK (Figure 4.4). The parent compound (1) demonstrated a P-gp efflux ratio of 5.8, and upon methylation of one of the sulfonamides (2) the ratio dropped to 3.2 (remember that < 2 indicates the molecule has limited P-gp efflux).
However, removal of the proton is not always tolerated and in such cases reduction of HBD character is beneficial. This can involve introducing an electron withdrawing group to reduce the HBD capacity of an amide/sulfonamide by removing electron density from the N-atom and modulating the pKa. For example, another series of bradykinin B1 receptor antagonists developed by Merck was shown to be substrates for P-gp. Careful manipulation of the alpha-amide substituents by appending fluorine atoms inductively reduced electron density from the amide and reduced P-gp efflux (Figure 4.5).
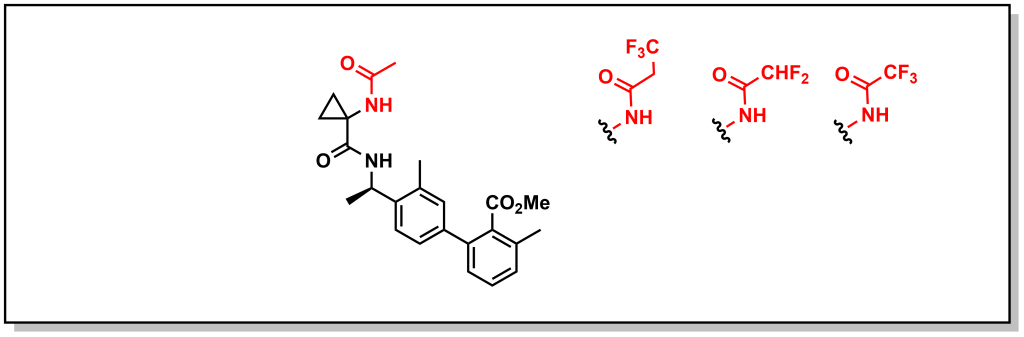
Other creative strategies involve masking the H-bond donor such as locking a pre-formed H-bond. For example, BACE (β-Amyloid cleaving enzyme-1) inhibitors are a target in Alzheimer’s disease that were pursued by Amgen. However, the amide proton was shown to facilitate P-gp efflux. Installing R-groups with a hetero-atom facilitating an intramolecular H-bond with the amide proton, masked the availability of the HBD thereby reducing P-gp efflux (Figure 4.6). For example, the ether linkage demonstrated correct positioning for intramolecular H-bond formation. Interestingly, the fluorinated pyridine showed reduced P-gp activity, which was not simply case of increased bulk around the H-bond (the non-fluorinated pyridine still demonstrated P-gp efflux). Instead, the fluorine atom of the pyridine is positioned to participate in an H-bond. This is a creative strategy since H-F bonding is weaker than H-O or H-N, but was structurally confirmed to participate in an H-bond and is successful at blocking P-gp activity.
4.3.4.1.2 Amines and Heterocycles
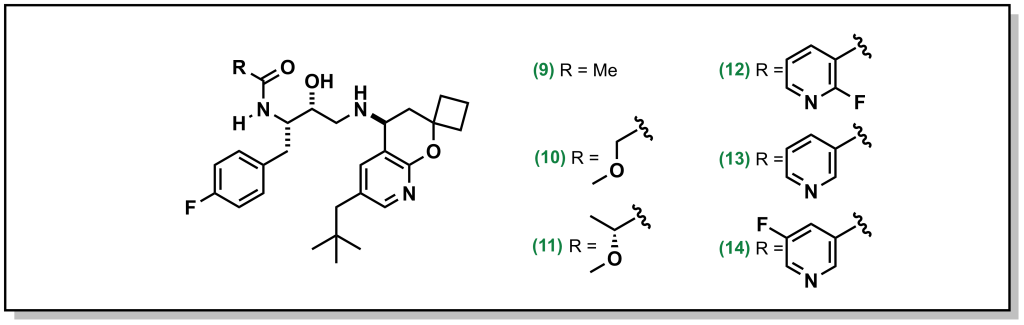
Basic amines and heterocycles can also impart P-gp efflux onto drugs, and reducing the pKa of the amine can improve activity (similar to above). For example, orexin antagonists developed by Merck for narcolepsy treatment contain imidazole analogues. Although the parent compound demonstrated an efflux ratio of 13, direct methylation of the imidazole removes the proton and dropped the efflux ratio to 2 (Figure 4.7).
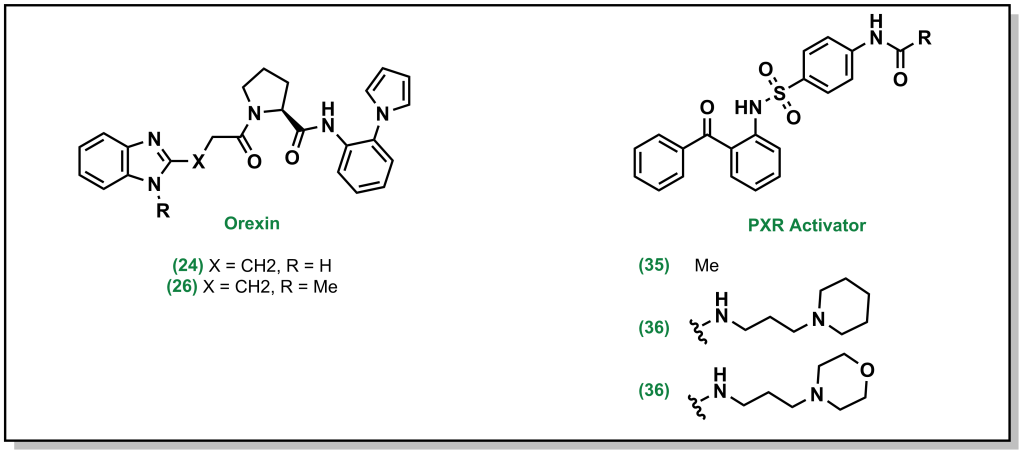
In cases where removing or blocking the heteroatom is not possible, reducing the pKa is also feasible. For example, the PXR activator was shown to have low metabolic stability which was improved by installing of the piperidine ring. However, the amine resulted in P-gp efflux, and switching to a morpholine ring inductively reduced the pKa of the nitrogen and subsequently the P-gp efflux (Figure 4.7).
4.3.4.1.3 Alcohols and Carboxylic Acids
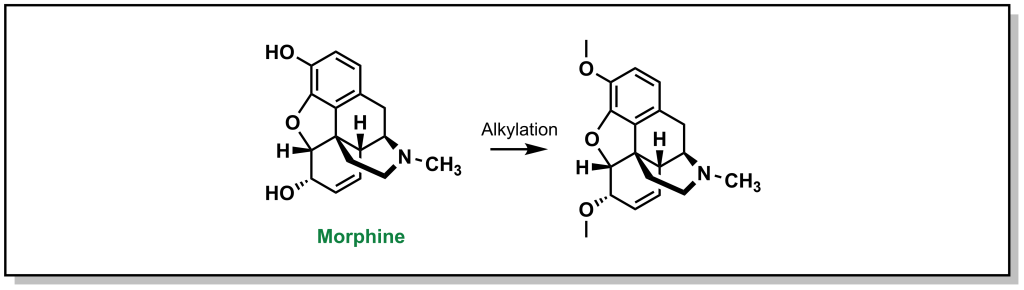
Alcohol and carboxylic acids follow the same principles outlined above, and the most common strategy is removing the proton through alkylation. For example, tolerance to morphine is thought be partially imparted by increased expression of P-gp (Figure 4.8). Likewise, carboxylic acids can be converted into other substrates functionalities such as esters, carboxamides, or dimethylformamides, which can also facilitate in masking the anionic charge and improving cellular permeability.
4.4 Absorption In Vivo
The above absorption parameters predominantly only consider cellular permeability. Within more complex living systems, additional factors need to be considered. For an in vivo experiment, the route of administration affects the rate of absorption and effective concentration as the drug is required to survive the journey through different organs and tissues of the body to find its target. Some of the different routes of administration are shown in Table 4.1. The main focus will be on IV, PO, and IP, although the FDA has over 100 approved administration routes.
4.4.1 Intravenous Administration
In intravenous (IV) administration, the drug is administered directly into the veins via a needle. This provides a rapid onset of action because the drug directly enters the blood stream without passing through any organs and there is no barrier to absorption. Drugs may be administered as a bolus (single dose) or infusion (continuous drip). Once the drug enters systemic blood circulation it is distributed throughout the body. A critical point of IV administration is that it avoids first-pass metabolism. This refers to metabolism of the drug at tissues in the body before it reaches its site of action. For example, remdesivir is an antiviral drug that is administered by IV injection. If it was administered orally, it would be subject to metabolism in the liver where it would be chemically altered before it had a chance to reach the intended site of action. Although IV administration allows for rapid onset, precise control of dosing, and avoids initial metabolism, it is not the preferred route for drug delivery because of the inconvenience and training required for administration of the drug.
4.4.2 Oral Administration
Oral formulations (pills, gels, liquids) are the preferred route, partly because of the ease of administration and patient compliance. However, the drug is taken by the mouth which means it passes through the stomach, small intestine (allowing absorption into the blood), liver, right atrium/ventricle of the heart, and then into full circulation. As such, there is generally a delay in onset as action, as the drug needs to pass through several organs before it enters systemic circulation. Furthermore, the drug must survive the acids/enzymes of the stomach, achieve absorption via the small intestine, survive metabolism in the small intestine and metabolism in the liver. Unsurprisingly, due to these hurdles in absorption and metabolism, not all of the drug that is administered orally will enter systemic circulation so oral doses are usually higher than IV doses. The amount of drug that reaches the bloodstream can be compared the amount of drug that enters the bloodstream if the patient was given the drug via IV administration, and this is represented quantitatively as %F which is called the bioavailability. A low %F indicates that the drug has poor bioavailability and is either poorly absorbed, rapidly metabolized or both, but does not always preclude a drug from therapeutic usefulness. The bioavailability of aspirin is ~50% and morphine as a bioavailability of ~30% as these drugs are metabolized in the liver. Warfarin is blood thinning agent whose %F is >99% which is exceptionally unusual for a drug.
4.4.3 Intraperitoneal Administration
Intraperitoneal administration is important in preclinical models and trying to isolate challenges in bioavailability. After injection, the drug is ultimately absorbed in the superior mesenteric vein (meeting up with other orally administered nutrients/drugs/substances that come from the small intestine capillaries via the jugular vein), which converges with the hepatic portal vein and enters the liver. This indicates that drugs administered via IP still undergo first pass liver metabolism, although absorption/degradation by the stomach and small intestine does not occur.
4.4.4 Pharmacokinetic Parameters
Quantitative pharmacokinetic profiling of a drug is usually done by analysis of the blood using mass spectrometry. Following administration of a specific drug dose (via any route), the blood is sampled at different time points. The sample is processed and analyzed by mass spectrometry to determine the concentration of drug in the blood at that time point. Plotting the concentration of compound over time allows for a number of important pharmacokinetic parameters to be determined:
- Cmax: This is the maximum concentration of drug in the blood that is achieved throughout the entire study. This provides an estimate of how well the drug is absorbed.
- Tmax: This is the time at which Cmax occurs. This provides an understanding of how long absorption takes.
- t½: This is the half-life which reports on the how fast the drug is eliminated from the blood.
- AUC: This is the area-under-the-curve which is the integral of the concentration of drug vs. time. This provides an estimate of the total exposure of the body to the drug and helps to put Cmax and t½ into perspective. Two potential drugs could have the same Cmax but if one candidate has a double the AUC value, that means this body has twice as much exposure to the drug.
Optimizing these parameters can vary between each drug. For example, using a drug that has a short-half life can be challenging, since it might be completed eliminated. However, if the drug has a very long half-life there can be toxicity side effects. Therefore, the pharmacodynamic effects also need to be considered when evaluating these parameters.
The PK profile for an IV administered drug usually demonstrates a Cmax at the initial timepoint since the drug is injected directly into the blood. As time passes, the drug distributes across different tissues and the blood concentration is reduced. For an orally administered drug, the drug has to pass through several organs before entering the blood stream and Cmax usually occurs later in the PK profile.
4.5 Distribution In Vivo
Following absorption, the drug enters systemic circulation and can access different parts of the body, including the site of the target. In the absence of any confounding variables (such as interactions with tissues or proteins), the drug would continue to circulate in the bloodstream and end up passing through the liver and kidneys with each cycle, and be successively metabolized or cleared until the drug is fully eliminated. However, a number of additional biological processes occur during distribution.
4.5.1 Whole Blood and Plasma Protein Binding
The first aspect to consider in distribution is the composition of fluid that harbours the drug, i.e. whole blood. Whole blood is a mixture of blood cells (red blood cells, white blood cells, and platelets that make up ~45% of the volume of blood) and plasma (~55% of whole blood volume). Plasma is fluid that contains water, electrolytes, and proteins (with the most common plasma protein as albumin at ~45 mg/mL). The high concentration albumin makes it a key target for engaging with drugs in circulation, and its common to have both a drug fraction-bound and drug fraction-unbound. The free-drug hypothesis states that the concentration of free-drug in the plasma (as opposed to total drug concentration) determines the therapeutic activity. Therefore, only the unbound fraction or free drug is available to carry out therapeutic effects. From this, it can be inferred that a drug that is administered and is highly bound to plasma proteins may not be as effective. For example, the blood thinning agent drug warfarin has an observed bound-fraction of 97% (this is actually favourable for warfarin since as a blood thinner, the predominant site of action for warfarin is the blood). Albumin is largely positively charged, and acidic or neutral drugs display a higher propensity for binding. Basic drugs can exhibit binding to the negatively charged alpha-1 acid glycoprotein.
Plasma protein binding can also lead to different effects that can further complicate drug distribution. For example, once the drug binds to plasma proteins, it act as a reservoir for drug: as drug reaches its target site and diffuses into the appropriate tissues, the concentration of free drug in the bloodstream will be reduced, and Le Chatelier’s principle indicates that bound-drug will be released to maintain a dynamic equilibrium. Alternatively, if a patient is administered two drugs that exhibit plasma-protein binding, these drugs may compete for the same binding sites on the plasma proteins. As such, the drugs may exhibit lower apparent plasma protein binding and this can lead to a higher effective dose (ie. unbound drug) realized in the patient, than if the two drugs were administered separately.
4.5.2 Volume of Distribution
In addition to distribution within whole blood, the drug will also partition into different tissues. This is often described through ‘compartment analysis’. The body can be thought of as different compartments, where a compartment refers to spaces/tissues with similar properties (and not necessarily separate organs). For example, blood can be assigned one compartment and all non-CNS tissues can be classified as a second compartment. Additional compartments can be incorporated depending on the extent of the distribution analysis.
The distribution of a drug is given by the apparent volume of distribution (Vd). A larger volume of distribution indicates the drug does not remain in the blood and enters other tissues or compartments. Conversely, a lower volume of distribution indicates the compound remains the blood. Therefore, a higher dose would be required for a drug with a high Vd to obtain the same plasma concentration as a drug with low Vd. For drug administration, Vd is an important parameter for to determine the dose to be applied to a patient.
Importantly, the Vd is not a real physical volume but provides an understanding of drug accumulation in the body. The average patient weight is 70 kg and the total volume of water in the body (~70%) is 49 L. Furthermore, there is approximately 5 L of blood per person, and since ~55% of the total blood is plasma, there are ~2.75 L of plasma in a patient. Based on these numbers, a drug with Vd > 40 L is thought to be distributed throughout all of the tissues in the body. Some drugs such as chloroquine (Vd = 15,000 L) or atorvastatin (Vd = 420 L) are well over the actual physical volume of the body. These high Vd values indicate that the compound highly concentrates in tissues in the body and does not remain within the blood. Chloroquine is an antimalarial drug that is highly lipophilic which helps explain its high Vd. Atorvastatin is also lipophilic and is known to accumulate in muscles which is partially responsible for some of its observed side effects. By contrast, warfarin and ibuprofen have low Vd values of 10 L and 10.5 L respectively, indicating they remain the blood. As the volume of distribution increases, the half-life of the drug also becomes longer. This is because the drug is distributed across more tissues which lead to additional time required for undergo renal or hepatic clearance.
