5 Drug Metabolism
“I can’t change the direction of the wind, but I can adjust my sails to always reach my destination.”
– Jimmy Dean
5.1 Biological Pathways for Drug Metabolism
Following administration, drugs are absorbed and distributed into different tissues. Over time, these molecules are eliminated (irreversibly removed) from the body which can occur in two ways: either directly unchanged (excretion/elimination) or changed (metabolized). Drug elimination predominantly occurs through the kidneys (especially for hydrophilic species) and exits via urine, although a subset of drugs (predominantly non-polar substances) are eliminated in the bile (via action of the liver) and excreted in the feces.
The liver is the principal site for drug processing and metabolism and has (without exaggeration) hundreds of characterized functions including roles in digestion, detoxification, immunity, hormone release, ammonia processing, glycogen production, and heavy metal storage. The liver also produces bile, which is a mixture comprised of sterol derivatives (bile salts), cholesterol, phospholipids, and electrolytes, that is stored in the gall bladder and released in the small intestine to facilitate digestion of fats. This hydrophobic character of bile also becomes important in the metabolism of highly non-polar drugs.
The liver holds approximately 10-15% of the total blood supply at any moment, and all blood that departs the stomach or intestines passes through the liver enabling absorption of nutrients (or metabolism of xenobiotics in the first-pass effect). The liver is comprised of functional units termed lobules, which are typically hexagonal structures characterized by a hepatic portal vein, artery, and bile duct at each hexagonal vertex. The portal veins drain into a central vein at the focal point of the hexagonal lobule, that carries blood away from the liver into systemic circulation. These veins (as well as the ducts and arteries) are lined by parenchymal liver cells which are called hepatocytes. Hepatocytes comprise approximately 60-70% of all the cells in the liver and have an asymmetric cellular structure with the periportal side (Zone 1) facing oxygenated blood/nutrients. The cellular face furthest away from the portal triad is called Zone 3 and plays the largest role in detoxification and biotransformation of drugs. The space sandwiched between these zones (pericentral region) is intuitively referred to as Zone 2 of the hepatocyte.
Importantly, the flow of blood through the veins and the flow of bile through the ducts occur in opposite directions to each other. This is consistent with the generation of both substances as bile is produced in the liver, whereas blood enters the liver for perfusion. As mentioned, blood drains from the branches of the hepatic veins into the centre vein of the lobule.
5.2 Phase I Metabolism
Drug metabolism refers to the chemical modification of a drug, and the overall goal is to generate a more polar or water-soluble derivative that is amenable to elimination. Drug metabolism is generally classified as either Phase I or Phase II, although these do not need to be successive steps, and some drugs will only be processed through one of these two phases of metabolism. Both phases of metabolism occur in the hepatocytes of the liver.
Phase I metabolism is characterized by functionalization of the drug via one or more of the reactions:
- Oxidation (electron removal, dehydrogenation, or oxygenation)
- Reduction (electron addition, hydrogenation, or removal of oxygen)
- Hydration/dehydration (hydrolysis, and addition or removal of water)
These transformations are carried out in the endoplasmic reticulum of hepatocytes and the most prominent Phase I reactions are oxidations. In an ex vivo setting, hepatocyte cells can be disrupted and processed by equilibrium density centrifugation to isolate vesicle-like pieces of the endoplasmic reticulum which are called microsomes. Although microsomes do not exist naturally, they provide a convenient tool to analyze Phase I metabolism. Over 75% of Phase I metabolism is performed by a class of enzymes called Cytochrome P450 (CYPs or P450) enzymes. These enzymes are heme-coupled monooxygenases and they bind oxygen (as well as carbon monoxide which leads to a wavelength absorption at 450 nm and serves as their namesake).
5.2.1 Cytochrome P450 Enzymes
There are at least 57 CYP enzymes encoded within in the human genome, with CYP3A4 and CYP2D6 having the highest abundance and contributing to >50% of all CYP-related metabolism. The nomenclature for these enzymes is based on the prefix CYP followed by an Arabic numeral indicating the protein family (usually >40% sequence identity), followed by a letter for the sub-family (>55% sequence identity), and the final number for the isozyme (often greater than 90% sequence identity). Importantly, genetic polymorphisms between CYP family members can result in differential processing of drugs between different genetic populations. For example, the drug diazepam (anxiety and seizure medication) is primarily metabolized by CYP3A4 and CYP2C19. However, mutations in 2% of Europeans, 14% of East Asians, and 57% of Oceanians result in deficient/absent CYP2C19 activity which leads to prolonged effects of the drugs in these patients. Poor drug metabolism can lead to an exaggerated pharmacological response, and the longer lifetime of the compound can also increase toxicity. Conversely, super-fast or rapid metabolizers (individuals with highly active CYP isoforms) can have altered drug responses by due to substantially lowered active drug lifespan. In both cases, clinical implications of polymorphisms require altering the dosages for the therapeutic regimen.
Since multiple drugs converge on CYP enzymes for metabolic processing, these oxygenases are also responsible for a number of drug-drug interactions. Several drugs are either substrates for CYP enzymes or act as inhibitors (prevent a substrate from engaging the CYP enzyme) or inducers (promote activity or gene synthesis of the CYP enzyme). For example, saquinavir (a protease inhibitor used for HIV treatment) is a substrate for CYP3A4, but can be administered with ritonavir (a CYP34A inhibitor) which enables a 33% increase in peak plasma concentrations of saquinavir. Similarly, serotonin uptake inhibitors such as fluoxetine inhibit CYP2D6. CYP2D6 processes codeine to morphine and a patient prescribed these inhibitors would therefore not benefit from the analgesic properties of codeine. There are also multiple inducers, for example CYP1A2 bio-transforms carcinogenic chemicals from cigarette smoke and is also induced by these agents in a positive feedback cycle. CYP profiling to determine which enzymes process a newly developed drug is important to determine any contradictions and metabolism concerns.
The mechanism for CYP oxidation is based on free radical chemistry that leverages different oxidation states of iron to facilitate electron transfer between the metal centre bound within a porphyrin ring called protoporphyrin IX (Figure 5.1). The overall process involves a drug (CYP substrate) with a potentially oxidizable bond (X-H) binding the low-spin Fe3+ (one unpaired electron S = ½, with a hexacoordinate metal centre including one water molecule) leading to a high-spin Fe3+ (five unpaired electrons S = 5/2, pentacoordinate with no water, and a shortened Fe-S bond , pulling the iron slightly below the plane of the heme). The drug substrate binding also triggers reduction of Fe3+ to Fe2+ with an electron originating from NADPH. The reduced Fe2+ has a high affinity for diatomic gasses such as O2 (and also CO) and transfers the electron to generate a superoxide Fe3+ complex. The Fe3+ ion picks up an additional electron (again originating from NADPH), which is also transferred to the superoxide anion to generate a peroxide anion (O22-) species that is split by two protons to form one water molecule and an iron-oxo complex called oxene. This oxene species is highly electrophilic and reactive and inserts into the substrate X-H bond yielding the desired oxidized product. This process only superficially describes the crystal field theory required for understanding these redox reactions, and the most relevant aspects for medicinal chemists are in understanding the types of C–H bonds that are susceptible to CYP oxidation and their final products.
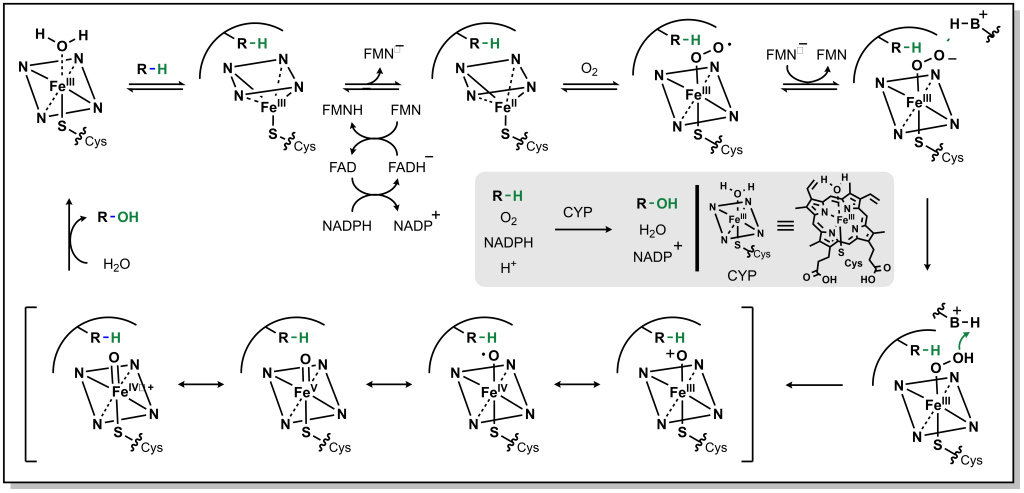
FIGURE 5.1 Reaction mechanism for metabolism via CYP enzymes.
5.2.2 Oxidation of sp3 Carbon Atoms
5.2.2.1 Aliphatic Straight Chain and Ring Systems
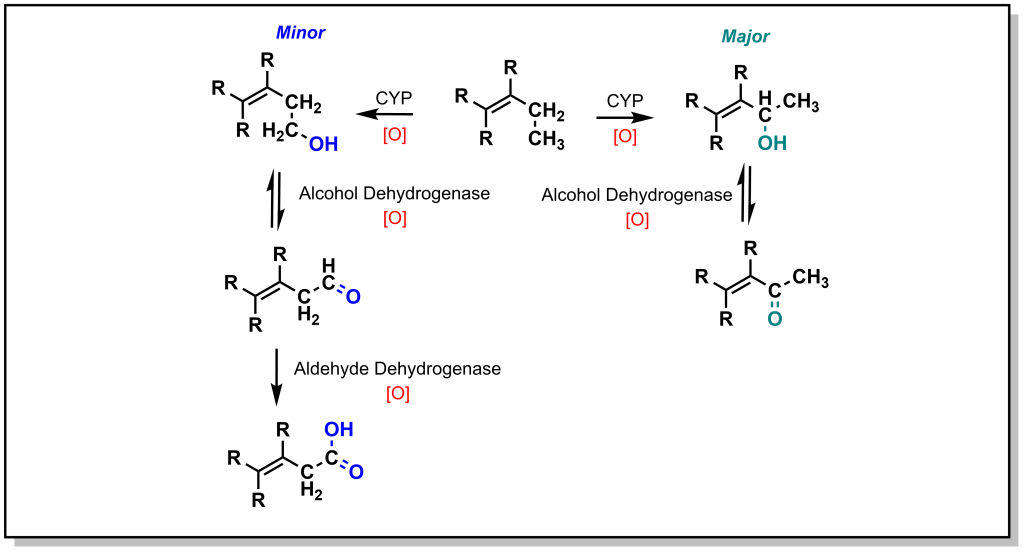
The main mechanism for metabolism of an sp3 carbon atom is hydroxylation. If there are multiple sp3 carbon atoms in the drug, there is a preference that hydroxylation will occur at the more substituted carbon atom. This site selectivity arises from the electronics of radical stability. Consider the homolysis of the C-H bond during cleavage in Figure 5.2. The radical generated on the carbon atom is most stable on the ‘internal’ carbon atom and forms the major product. Regardless, the alcohol generated from both the major and minor products will be further oxidized by a non-CYP Phase I enzyme called Alcohol Dehydrogenase (ADH) to give a carbonyl (ketone or aldehyde). For any aldehydes generated as a result of terminal carbon oxidation, the molecule is further oxidized by a comparable Aldehyde Dehydrogenase (ALDH2) to yield a carboxylic acid. Notably, alcohol dehydrogenation is reversible, but the activity of aldehyde dehydrogenase is not reversible.
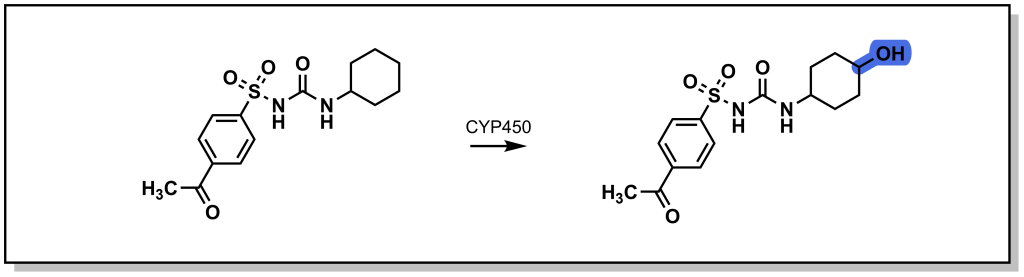
Saturated ring systems are a common functionality in different drug molecules. Due to sterics, oxidation generally occurs at the 3- or 4- ring position as shown with the metabolism of acetohexamide in Figure 5.3. Substitution at the 4- position during SAR design is often considered to impede Phase I enzyme access and sterically block hydroxylation, thereby extending drug half-life.
5.2.2.2 Allylic and Propargylic Systems
As shown with both examples above, both sterics and electronics can play a role in metabolism at sp3 carbon atoms. As such, there is a site preference of hydroxylation of sp3 carbon atoms alpha to an unsaturated system. Based on electronic effects, the proximity to a double/triple bond provides stability to a radical (Figure 5.4). Furthermore, in an unsaturated system, the resulting alcohol is dehydrogenated to create a 1,4-conjugated system which has excellent electrophilic properties for Phase II metabolism.
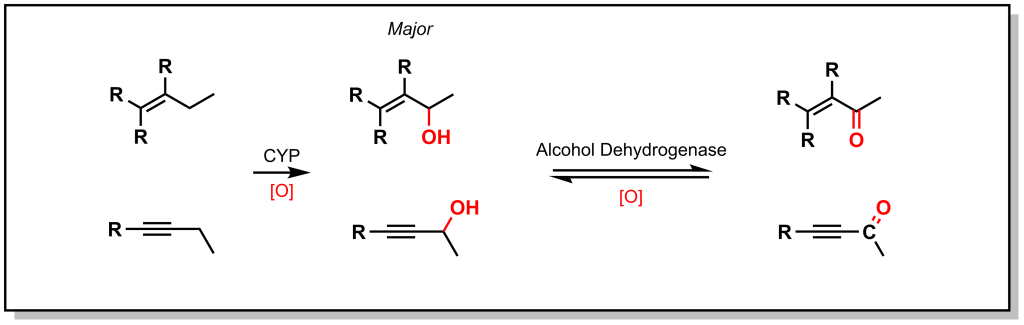
Hexobarbital (a barbiturate derivative used as an anesthetic in the mid-20th century) contains a cyclohexene substituent (Figure 5.5). Based on sterics, metabolism could be favoured at the 4-position. However, hexobarbital is hydroxylated by CYP2B1 at the 3- position, alpha to the double bond (due to increased electronic stability of the radical). The resulting alcohol is either oxidized to an α,β-unsaturated ketone or is a substrate for UDP-glucuronosyltransferases (discussed in later sections on Phase II metabolism).

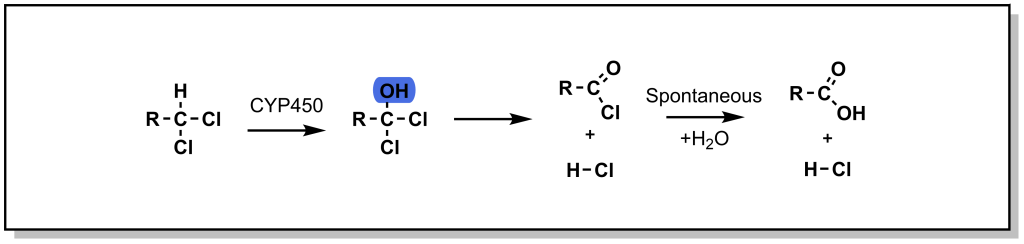
5.2.2.3 Halogenated Systems
In the case of a halogenated sp3 centre, oxidation (hydroxylation) can lead to a high-energy / unstable intermediate, that can undergo elimination of the halogen to form a ketone. This oxidative dehalogenation is more prevalent in the presence of two halogen atoms on the sp3 centre. For example, chloramphenicol is an antibiotic that disrupts bacterial protein synthesis. Chloramphenicol possesses a terminal di-chloro moiety that is oxidized to an acid halide intermediate (very reactive) and further reacts to form a terminal acid moiety. Note that oxidative dehalogenation is not feasible with fluorinated carbon atoms, as the C-F bond is extremely strong and cannot be homolyzed by CYP enzymes. In this way, fluorine can function as a bioisostere for hydrogen.
5.2.3 Oxidation of sp2 and sp Carbon Atoms
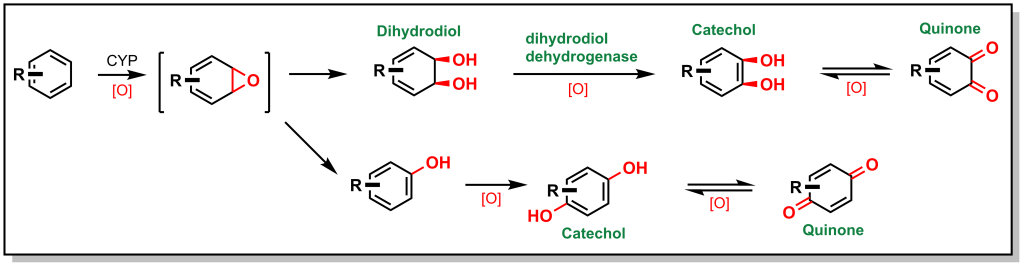
Unsaturated carbon scaffolds are also susceptible to oxidation by CYPs. For example, the oxygen atom from the CYP enzyme can insert into the π system of benzene to form a highly reactive epoxide (Figure. 5.7). The reactive epoxide is subsequently hydrolyzed by epoxide hydrolase to yield a dihydrodiol or it can re-arrange and re-aromatize to phenol via proton catalysis. In either case, these substrates are further oxidized to catechol (bis-phenol) derivatives. These catechols are substantially more polar and can be directly excreted, or oxidized by dehydrogenases to yield reactive quinones (which are substrates for Phase II metabolism).
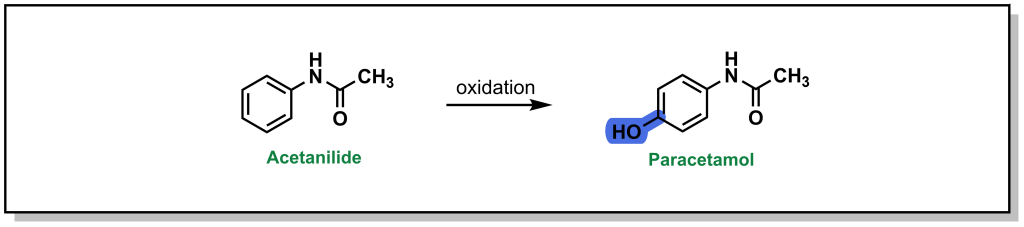
Acetanilide is a discontinued drug that was the first aniline derivative identified to possess analgesic properties. These favourable therapeutic properties are a result of aromatic hydroxylation at the para-position (sp2 carbon) which gives paracetamol (commonly known as acetaminophen or Tylenol). (Figure 5.8) However, there is also a minor metabolic product of N-dealkylation (discussed below) which results in aniline and is nephrotoxic leading to hemolytic anemias and other safety concerns.
Compared to sp3 oxidation, the metabolic modification of sp2 and sp carbons is a significantly slower process. Furthermore, it is also uncommon in modern drug discovery to observe free, unsubstituted phenyl rings in drug structures. This is partly because common strategies for bypassing sp2 metabolism involve generating more electron deficient rings or adding a fluorine or chlorine/substituent to the phenyl ring.
The utility of benzene in synthetic organic chemistry represents an interesting example of differences in drug metabolism of sp3 and sp2 carbons. As a simple aromatic liquid, benzene was routinely employed as an organic solvent for a number of industrial applications as well as organic synthesis. However, it is highly toxic and has been largely superseded by toluene, which still has safety concerns, but is far less toxic than benzene. This difference arises from the sp3 benzylic position of toluene which is much weaker (C-H bond strength = 85 kcal/mol) than the aryl C-H bond (110 kcal/mol). As such the benzylic carbon is more readily oxidized by CYP enzymes, creating a benzylic radical that is further dehydrogenated and oxidized to afford benzoic acid, which is rapidly cleared. Conversely, oxidation of the benzene sp2 carbon is substantially slower, and therefore benzene remains in the body for longer time periods. Moreover, the toluene oxidation metabolite has limited safety challenges, whereas benzene oxidation leads to a reactive quinone electrophile which is an excellent 1,4-Michael acceptor.
5.2.4 Oxidation of Hetero-atoms Bound to Carbon Atoms
Hetero-atoms (O, N, and S) are common in drug discovery and are often responsible for key target-drug interactions. The general strategy employed by Phase I metabolism enzymes is to either i) oxidize the hetero-atom directly (referred to as heteroatom oxygenation) or ii) oxidize the alpha-carbon next to the hetero-atom (often referred to as de-alkylation although there is an initial hydroxylation step; Figure 5.9).
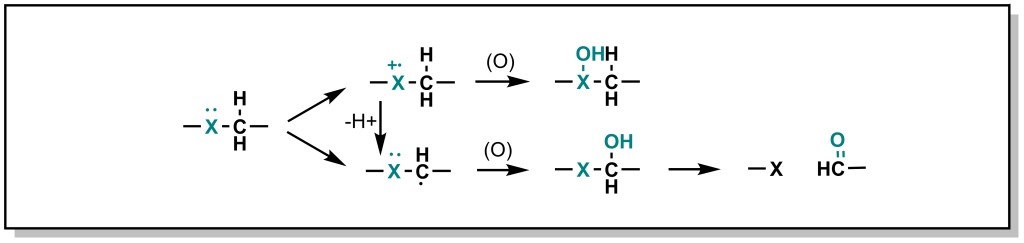
In heteroatom oxygenation, there is an initial single electron transfer (SET) from the heteroatom to the iron-oxygen complex of the CYP enzyme. This loss of an electron creates a radical cation intermediate on the heteroatom that eventually reacts with an oxygen provided by the CYP enzyme. In de-alkylation, there is an initial hydrogen atom abstraction (HAT) from the alpha-carbon which ends up on the iron-oxygen complex of the CYP enzyme (becoming iron-hydroxyl). This loss of a hydrogen atom creates a neutral carbon radical intermediate that eventually reacts with the hydroxyl provided by the CYP enzyme. The type of metabolism (heteroatom oxygenation or de-alkylation via SET or HAT respectively) that occurs depends on i) the hetero-atom and ii) the substituents of the heteroatom.
- For carbon-oxygen systems, heteroatom oxygenation does not occur because removing a single electron to generate a positively charged oxygen is highly energetically unfavorable. Therefore, for carbon-oxygen systems, metabolism occurs via HAT and de-alkylation.
- For carbon-nitrogen systems, HAT and dealkylation are highly favoured (similar to carbon-oxygen systems above). However, SET is not as unfavourable for nitrogen species and therefore heteroatom oxygenation is feasible and can proceed (usually as a minor product). SET/Hetero-atom oxidation is more favorable when the radical cation is destabilized based on the nearby substituents, or if there are no alpha-protons available for abstraction.
- For carbon-sulfur systems, SET and heteroatom oxygenation is energetically favored due to the formation of a stable sulfur radical cation.
5.2.4.1 Metabolism of O-Containing Moieties
5.2.4.1.1 Metabolism via O-Dealkylation
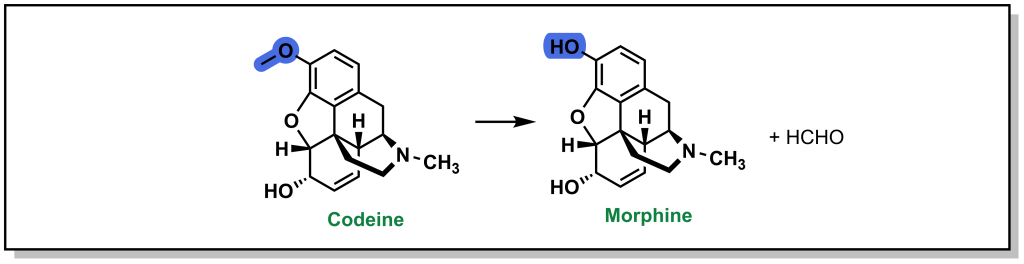
As mentioned above, the carbon-oxygen containing moieties will only undergo de-alkylation and will not be directly oxidized on the oxygen atom. These C-O moieties are commonly found as ethers in drugs and will metabolize to an alcohol with release of a ketone or aldehyde. As with all metabolic reactions, steric hindrance of the ether can block metabolism. A well-known example of O-dealkylation involves metabolism of codeine to morphine where the terminal ether is metabolized and also releases the parent phenol (Figure 5.10). Notably, this only accounts for ~10% of the modification of ingested codeine. Approximately 10% of codeine is also N-dealkylated (which will be discussed in the next section), and the remaining 80% is directly glucuronidated on the cyclohexenyl alcohol, in Phase II metabolism. Notably, the alternative C-O linkage in the ring system is not metabolized due to steric hindrance.
5.2.4.2 Metabolism of N-Containing Moieties
5.2.4.2.1 Metabolism via N-Dealkylation
The most common types of nitrogen containing compounds in drug discovery are tertiary amines (including both aliphatic and aromatic amines). Oxidation of these amines predominantly occurs as discussed above, where a carbon atom alpha to the nitrogen is hydroxylated to form a carbinolamine and this intermediate rearranges via cleavage of the C-N bond (de-alkylation; Figure 5.11).
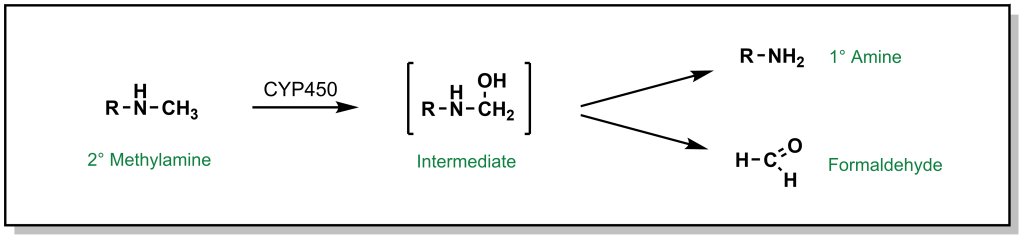
FIGURE 5.11 Oxidation of secondary amines can lead to N-dealkylation.
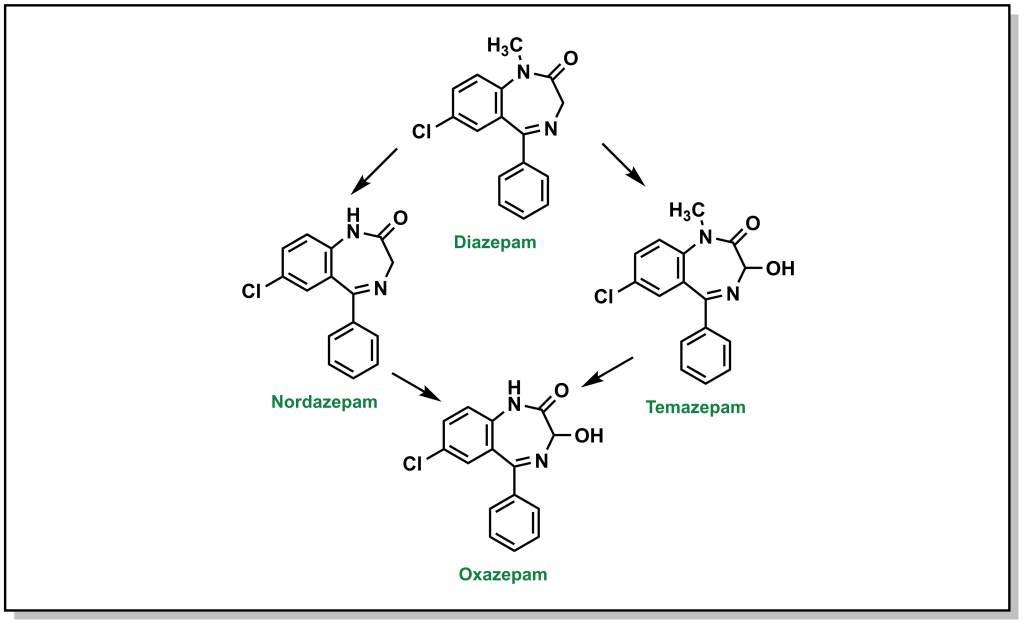
For example, the tertiary amine of diazepam is N-dealkylated in Phase I metabolism (Figure 5.12). Additional metabolic steps of diazepam include hydroxylation of the sp3 carbon which becomes an available substrate for glucuronidation.
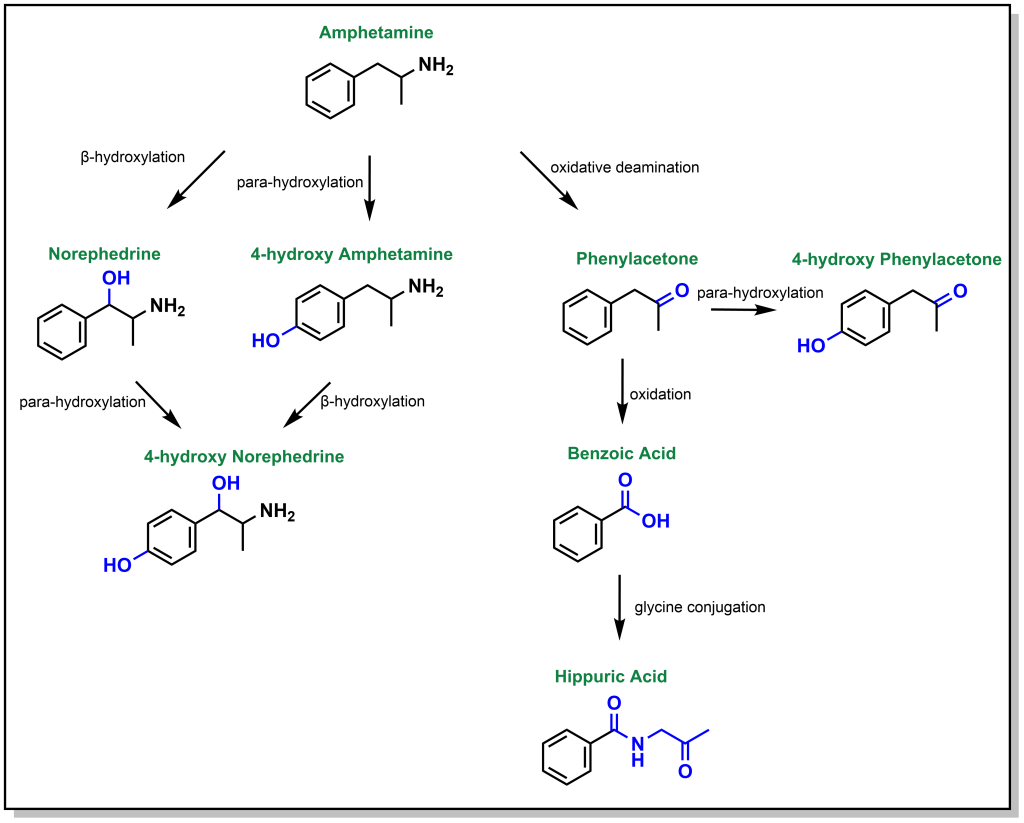
Dealkylation occurs for tertiary and secondary amines. Primary aliphatic amines are metabolized via the same mechanism, where hydroxylation of the alpha-carbon occurs, followed by rearrangement to release ammonia, and this is termed de-amination (as opposed to N-dealkylation). Amphetamine is a respiratory and CNS stimulant that is metabolically processed by different reactions including an oxidative deamination but also β-hydroxylation (of the sp3 benzylic carbon) and para-hydroxylation (of the sp2 phenyl ring; Figure 5.13).
5.2.4.2.2 Metabolism via N-Oxidation
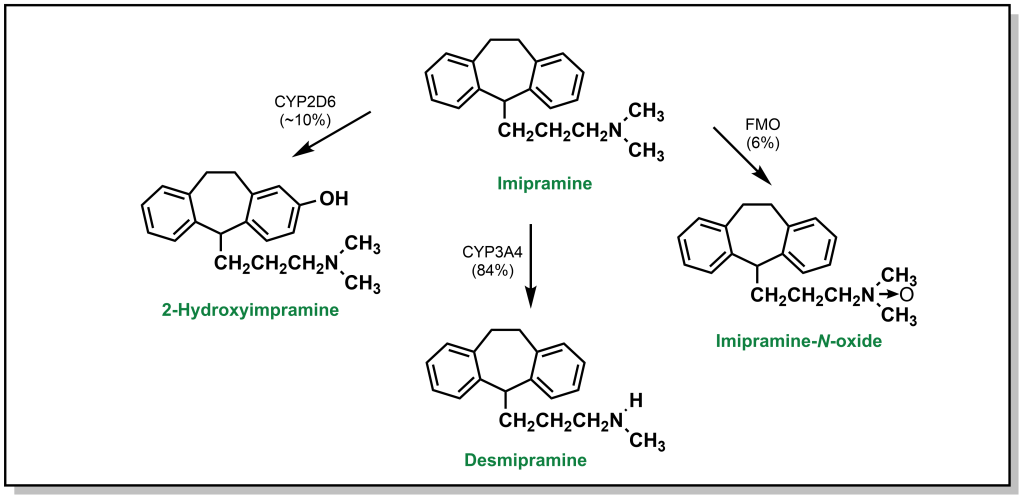
In addition to de-alkylation and de-amination, tertiary amines can undergo metabolic N-oxidation by a group of Phase I enzymes called flavin-containing monooxygenases (FMO) which enable oxidation on the heteroatom to yield N-oxide products. As mentioned, this is generally less favourable due to initial formation of a radical cation on the nitrogen atom. For instance, imipramine is an amine-based tricyclic antidepressant. Its tertiary amine is predominantly processed by hepatocytes through N-dealkylation via CYP3A4 (84%). Para-hydroxylation by CYP2D6 accounts for ~10% of the metabolic products observed. Finally, the remaining 6% is metabolized to an imipramine-N-oxide via FMO (Figure 5.14).
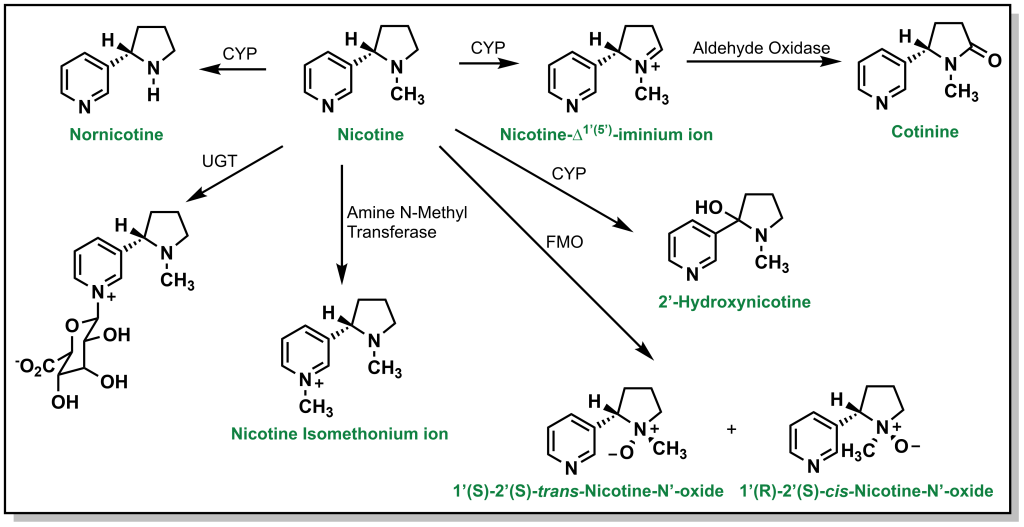
N-Oxidation can also occur with alicyclic amines such as nicotine, which harbours a nitrogen in the pyrrolidine and pyridine rings. Approximately 10% of nicotine is N-dealkylated at the pyrrolidine and ~75% is oxidized to cotinine (amide) via a similar hydroxylation mechanism. The pyridine can also be directly glucuronidated through Phase II processes (5%). Additional metabolism occurs through FMOs to create nicotine n-oxide (Figure 5.15).
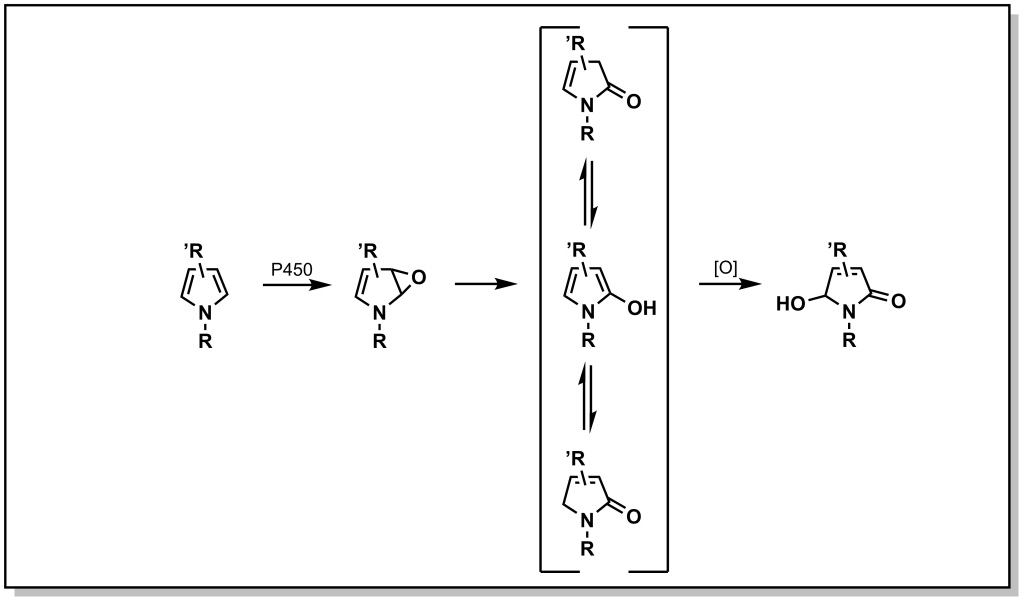
Nitrogen–containing aliphatic, and heteroaromatic ring systems are extremely common substructures in many drugs. Indoles and pyrroles represent electron-rich heterocycles which helps to facilitate metabolism via ring oxidation, with a preference towards oxidation on carbon atoms adjacent to the nitrogen. Metabolism trends from the analysis of several drug libraries indicate that a greater number of nitrogen atoms in the ring, reduce the possibility of Phase I oxidations. Tetrazoles in particular, are relatively inert to oxidation (and are important bio-isosteres of carboxylic acids, discussed later). Importantly, a number of nitrogen containing heterocycles including pyridines, imidazoles, and triazoles can engage with the heme-group to block activity of CYP enzymes. However, they can also be excellent substrates for Phase II metabolism, especially N-glucuronidation as observed with nicotine (Figure 5.16).
5.2.4.3 Metabolism of S-Containing Moieties
5.2.4.3.1 Metabolism via S-Oxidation
Phase I metabolism for sulfur containing compounds generally proceeds via direct oxidation of the heteroatom (SET), which impacts different sulfur containing functional groups including sulfides (especially in ring structures), free thiols, sulfonyls, and thioketones. Free thiols are uncommon in modern drug molecules due to their reducing potential, which enables them to form di-sulfide bridges with cysteine residues or self-aggregate. However, in such drugs, the thiol is directly hydroxylated to form sulfenic acid, which is further oxidized to sulfonic acid (Figure 5.17).
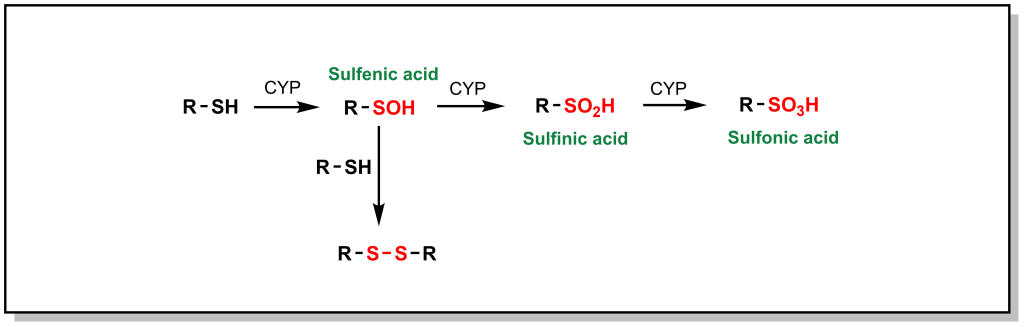
Higher oxidation states of sulfur, such as sulfonyls found in sulfonylureas and sulfonylamides, are significantly more hydrophilic, which facilitates their elimination. Thioethers (sulfides) can be oxidized to the corresponding sulfoxide and then further oxidized to the sulfone. For example, thioridazine (an antipsychotic medication used for schizophrenia) contains two sulfides and both of these sulfides can be S-oxidized (additional metabolic pathways include N-dealkylation; see Figure 5.18a). Thioketones undergo desulfuration via a similar oxidation of the sulfur atom as seen with thiopental, which produces a carbonyl-containing product (Figure 5.18b).
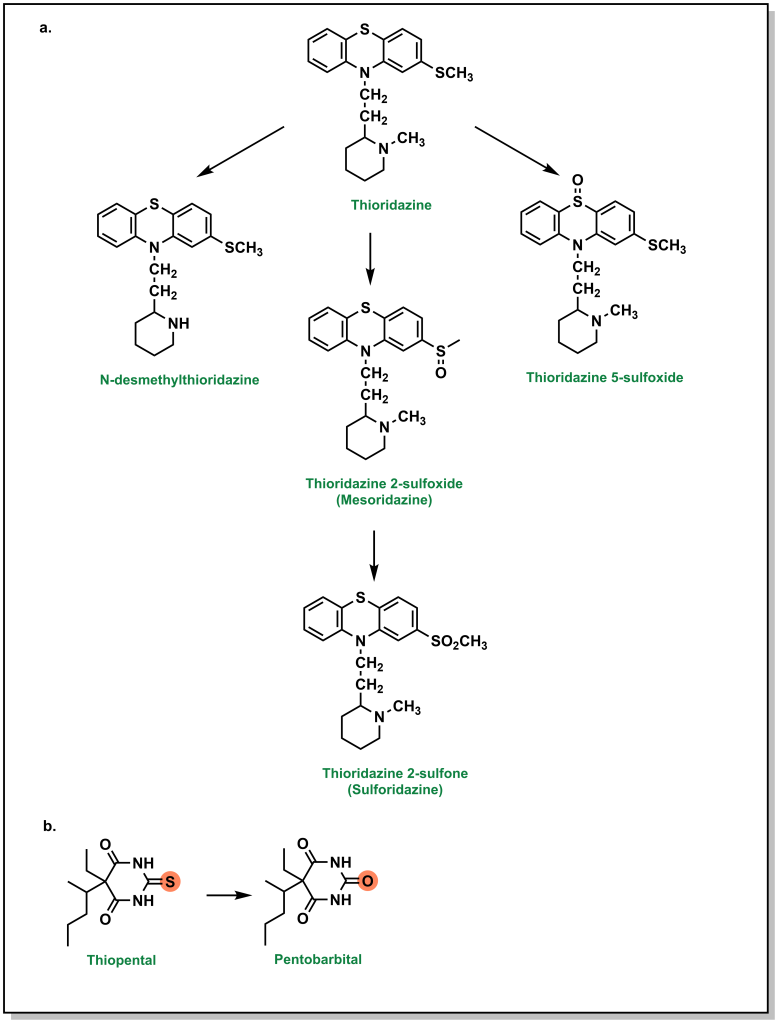
Importantly, metabolism of sulfur atoms has been associated with bioactivation and generation of reactive metabolites that are associated with toxicities, and understanding metabolite profiles can be important when selecting sulfur functional groups.
5.2.5 Other Phase I Oxidation Enzymes
In addition to CYPs, there are other non-CYP enzymes involved in Phase I metabolism, some of which have been highlighted above. The most commonly known non-CYP Phase I enzyme is Alcohol Dehydrogenase, which oxidizes alcohol functional groups into aldehydes or ketones, and Aldehyde Dehydrogenase, which oxidizes aldehydes to carboxylic acids. These enzymes are found exclusively in hepatocytes and have well characterized functions on ethanol, methanol, and ethylene glycol. Other important enzymes are Flavin Containing Monooxygenases (FMOs), which operate in a similar manner to CYPs and yield highly similar by-products. However, while CYPs utilize the high-reactive oxene as an oxidizing agent, FMOs use slightly less powerful peroxides. As such, FMOs can only oxidize heteroatoms and cannot oxidize primary amines, highly charged or polyvalent species.
Regardless of the oxidation mechanism, the overall goal of Phase I enzymes is to increase solubility (hydrophilicity) or expose a functional group for conjugation in Phase II.
5.2.6 Reductions
5.2.6.1 Carbonyls
Reductions are less common since they usually mask reactive groups, but they are largely involved in reducing carbonyls (mainly ketones) to hydroxy groups, which are more susceptible to conjugation reactions (Phase II). The order for reactivity generally follows the principles of sterics and electronics, namely aliphatic ketones/aldehyde > aromatic (benzylic) ketones/aldehyde > esters, acids, and amides. Importantly, the reduction of ketones can lead to the generation of stereocentres and stereoisomeric alcohols. For example, one of the routes for the metabolism of the blood thinner warfarin involves the reduction of its ketone by Human Hepatic Cytosolic Reductase to yield two diastereomeric alcohols.
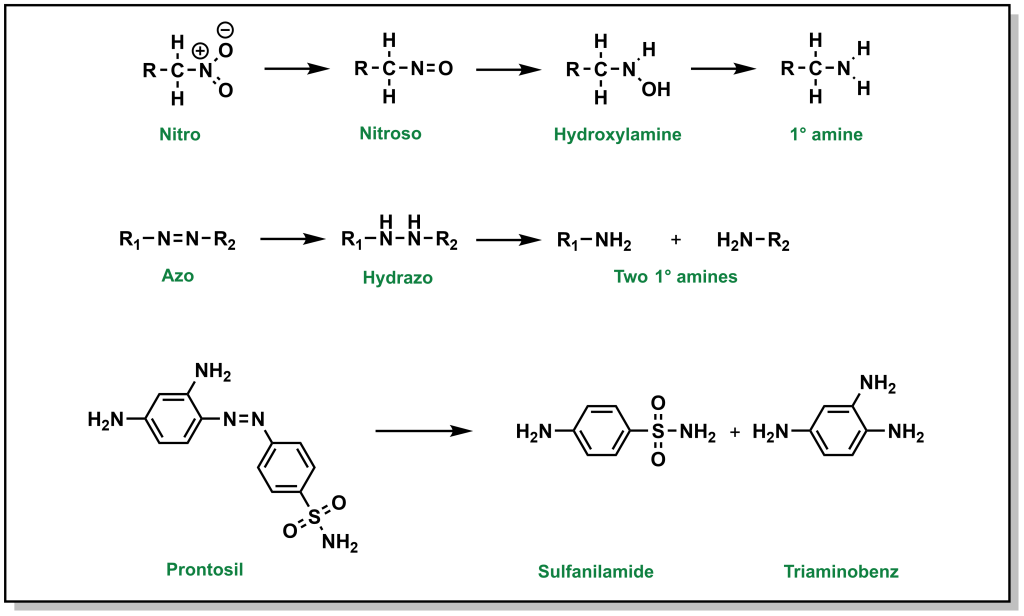
5.2.6.2 Nitro- and Azo- Compounds
Nitro- reductions are performed by NADPH-dependent microsomal and soluble nitroreductases, whereas NADPH-dependent multicomponent hepatic microsomal reductase systems reduce azo- groups. The end products of these reactions are primary amines (Figure 5.19). Bacterial reductases in the gastrointestinal tract have the capacity to reduce both nitro and azo groups.
5.2.7 Hydrolysis
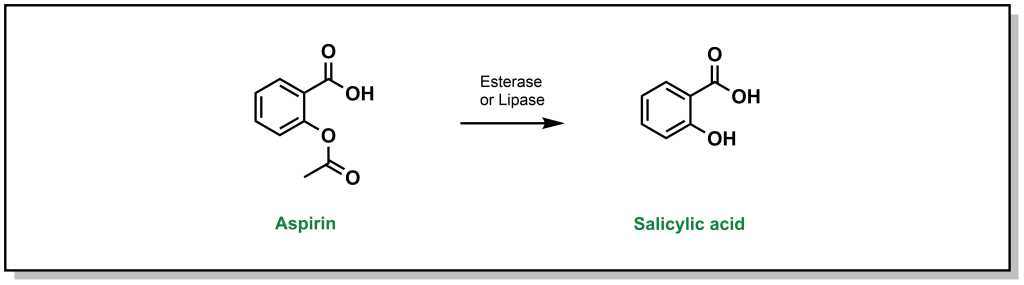
Hydrolysis reactions are fundamentally distinct from previous Phase I reactions described above, in that there is no change to the redox level of the drug molecule, and there is usually a substantial change to original substrate (cleavage of a functional group and generation of multiple products). The reactivity of a substrate towards hydrolysis is determined by the electrophilicity of the carbonyl carbon (or other reactive centre) and the nucleofugality of the leaving group substituents. The rate is found to follow the trend of thioester > ester > carbonate > amide > carbamate. One of the classic examples is hydrolysis of aspirin which can be converted to salicylic acid by esterases. (Figure 5.20)
5.3 Phase II Metabolism
Whereas Phase I is largely focussed on oxidases that aim to introduce a polar functional group, Phase II involves transferases that conjugate a biomolecule onto the drug. Phase I reactions may not necessarily generate a pharmacologically inactive species, but Phase II reactions are regarded as detoxifying and will inactivate drugs to prevent damage to macromolecules or to prepare drugs for efficient elimination. Substrates are covalently coupled to an endogenous molecule, which is generally very polar and large (100-300 Da) and can usurp the physical properties of the drug (LogP, tPSA, etc.). Since these processes can be energetically unfavourable, Phase II transferase enzymes will activate the endogenous molecule to prepare it for conjugation to the drug.
5.3.1 UDP-Glucuronosyltransferases (UGT)
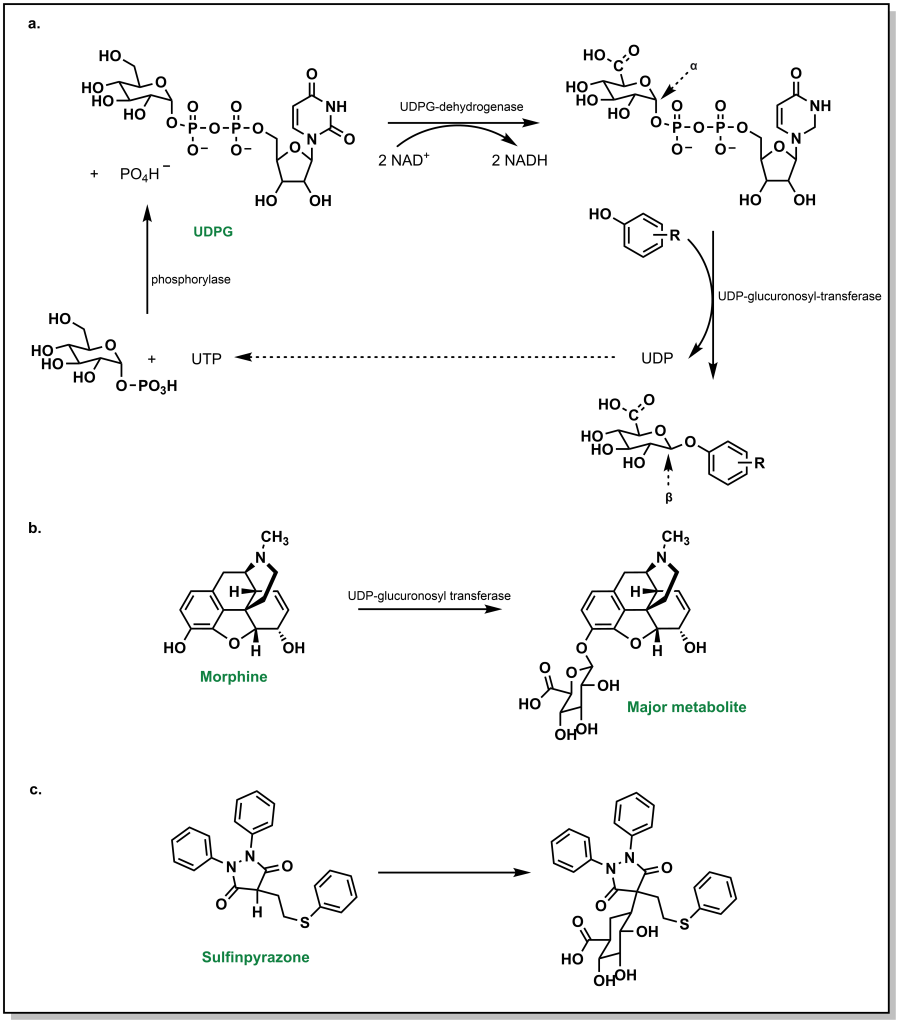
Glucuronidation by UGTs represents the most common Phase II metabolic process and involves the transfer of a glucuronosyl group to either an O-, N-, S– or C– atom. Physiologically, the ubiquity of glucuronidation arises from the availability of D-glucuronic acid (a D-glucose derivative), the enzymatic activation of the conjugate, and the stark changes in water solubility by addition of the glucose-group. In this conjugation reaction, glucose-1-phosphate is first activated to uridine-5-diphosphoate-alpha-glucose (UDPG) by the action of a phosphorylase. The 6-C is oxidized to an acid via UDPG-dehydrogenase and two equivalents of NAD+ (Figure 5.21a). The glycosidic bond is a high energy bond and easily broken to conjugate onto the substrate via nucleophilic attack of the drug substrate. This results in uridine diphosphate (UDP) functioning as a leaving group. Notably, this SN2 attack of the drug substrate also leads to inversion of stereochemical configuration in the resulting β-linkage to the glucuronic acid. A number of functional groups can participate in this reaction, including weak nucleophiles, and this accounts for approximately one-third of all Phase II metabolism. It is very common to form O-glucuronides via a hydroxy groups (phenolic/alcoholic) or carboxylates (aromatic or aliphatic). For example, morphine is extensively metabolized via O-glucuronidation (Figure 5.21b).
Even carbon atoms, in a minority of cases, can be glucuronidated such as in the case of sulfinpyrazone (a drug for the chronic treatment of gout). This occurs because the H-atom of the tertiary carbon is acidic and susceptible to removal by a base to create a nucleophilic carbanion that undergoes reaction with UDP-glucuronic acid (UDPGA) (Figure 5.21c). As described in previous examples, N-atoms and S-atoms can also be glucuronidated.
5.3.2 Glutathione-S-Transferase
Glutathionylation is the second-most common Phase II metabolism pathway, and also forms a significant aspect of first-pass metabolism. Glutathionylation is thought of as a highly protective process with de-toxifying effects. Glutathione is a tri-peptide analogue of glycine, cysteine, and glutamate, with a γ-peptide linkage between the carboxyl side chain of glutamate and the alpha amino group of cysteine. The reactivity of glutathione emerges from the cysteine side chain, which has an expected pKa of 8.7 (more acidic than a similar alcohol functionality such as serine, due to stabilization of the negative charge on a larger, polarizable sulfur atom). However, the nucleophilic character can be further enhanced by the action of the enzyme Glutathione S-transferase, which can increase the acidity by 2-3 pKa units, generating a charged thiolate anion. Any drug that has mildly electrophilic sites will be vulnerable to nucleophilic attack. The most common substrates of glutathione-S-transferases are soft electrophiles/Michael acceptors.
- α,β-unsaturated carbonyls (most common electrophile)
- quinones/quinone imine (often formed on Phase I metabolism)
- epoxides
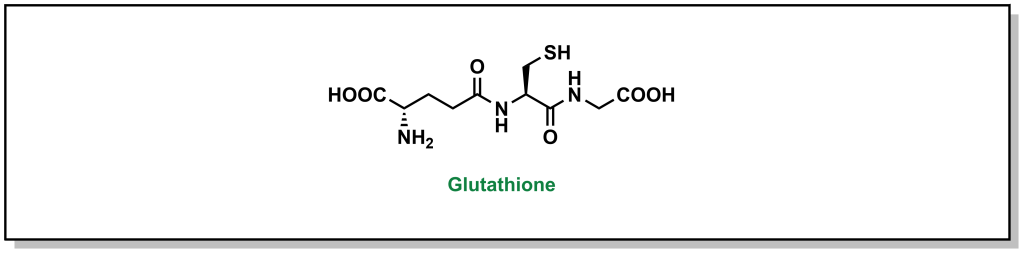
FIGURE 5.22 The tri-peptide glutathione.
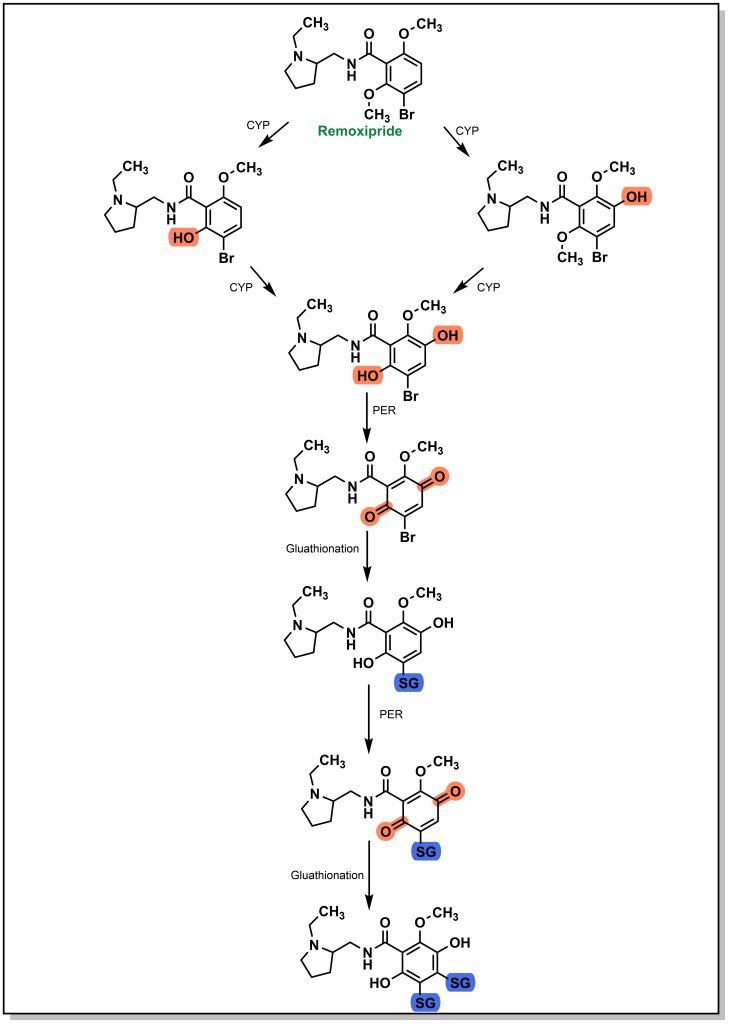
There have been a number of drug examples discussed above that are subject to glutathionylation, such as paracetamol (Tylenol). One case involves remoxipride, which is a substituted benzamide and was previously employed as an antipsychotic for treatment of schizophrenia. As expected, initial CYP metabolism leads to dealkylation of an ether linkage that is further oxidized to a quinone species (Figure 5.23). The presence of a bromo-group enables two glutathione transfer reactions, which inactivates the molecule and prepares it for elimination. Remoxipride was discontinued after 8 cases (2 fatal) of aplastic anemia were detected.
5.3.3 Sulfonyltransferases
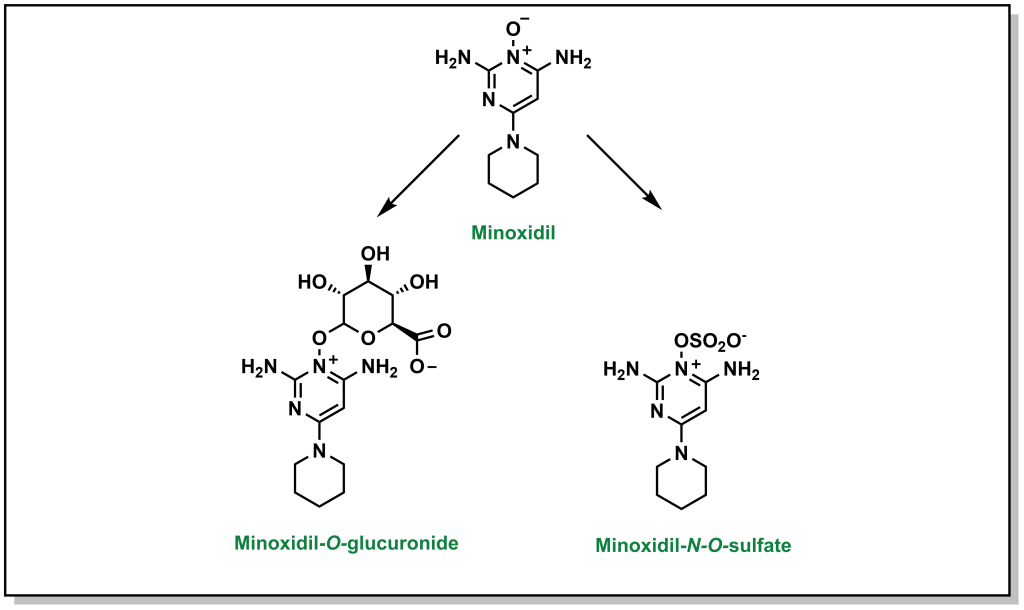
Sulfonylation involves the transfer of sulfate from an endogenous biomolecule to a drug (containing a nucleophilic hetero-atom). Similar to the above Phase II reactions, the introduction of a sulfate markedly alters the hydrophilicity. The sulfate group is provided by a molecule called 3′-phosphoadenosine 5′-phosphosulfate (PAPS) which contains a mixed sulfate/phosphate anhydride. This is a high energy anhydride that is significantly more reactive than di- or tri-phosphates, but has significantly reduced cellular availability compared to glutathione or UDP-glucose. This reaction predominantly occurs with phenols, but is known to occur with alcohols, aryl amines, and N-oxide compounds. For example, minoxidil (Rogaine) is drug used to help with hair loss in men and can be metabolized by the action of sulfonyltransferases (as well as by glucuronidation; Figure 5.24).
5.3.4 Methyl Transferases
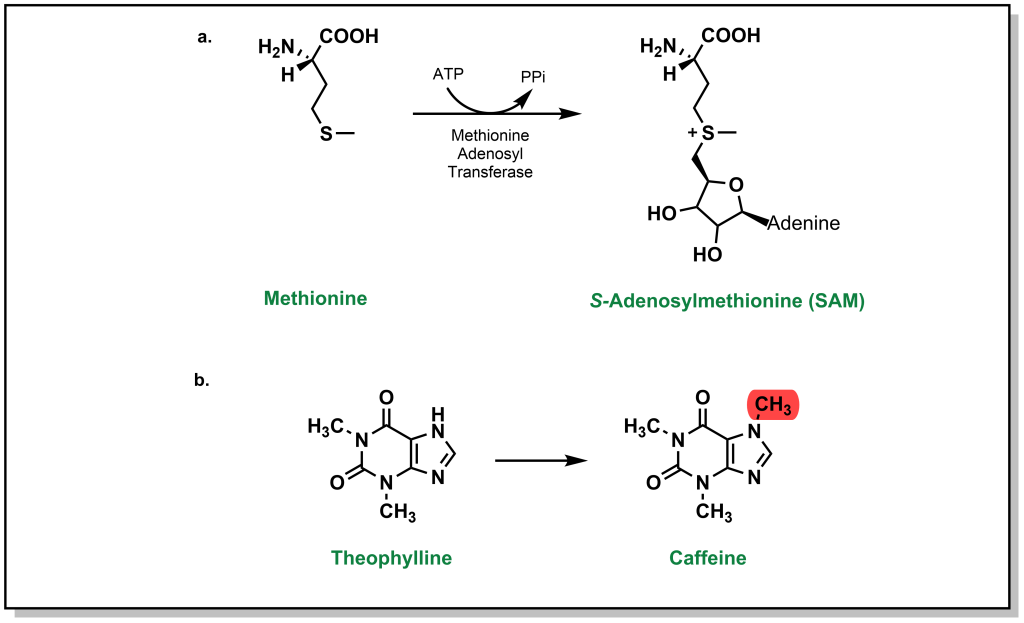
While glucuronidation, glutathionation, sulfonylation, as well as several other Phase II metabolism pathways increase the water-solubility of drugs, processes such as methylation (and acetylation below) do not substantially alter values such as LogP. Instead, these processes are tailored more toward inactivating a drug (an exception to his occurs with the formation of a charged, quaternary ammonium derivative). Any heteroatom with a hydrogen atom is prone to methylation, and this usually occurs at a late stage in the metabolism process. Methylation is carried out by S-adenosylmethionine (SAM) which involves an ATP-activated methyl group from methionine). For example, theophylline is used to treat apnea and breathing issues (which can be common in infants, especially premature babies), but the methylated metabolite is caffeine, which can lead to adverse effects (Figure 5.25).
5.3.5 Acetyl Transferases
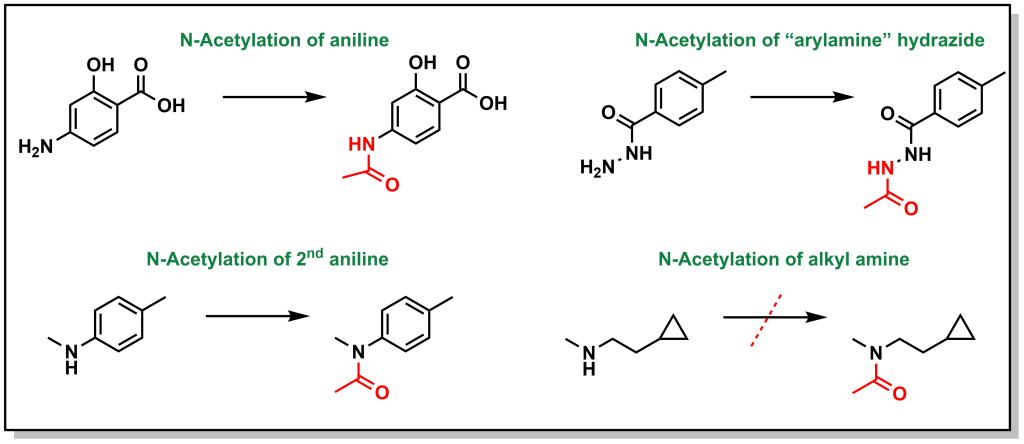
Similar to methylation, acetylation does not substantially alter lipophilicity but facilitates drug inactivation particularly on nucleophilic atoms (N and O). The main substrates are arylamines and sulfonamides, and to a lesser extent hydrazines, hydradizes, and aliphatic amines. The major enzyme system that acetylates the drugs is arylamine N-acetyltransferase. Primary and secondary aliphatic amines are only marginally acetylated (Figure 5.26). This reaction is particularly important in sulfonamide metabolism because acetyl-sulfonamides have reduced solubility and can lead to precipitation, especially in the kidneys, leading to renal toxicities
5.4 Drug Metabolism Exploits Ubiquitous Themes in Chemistry
One of the remarkable features of hepatocytes and drug metabolism within the body is that there is a high efficiency and capacity to metabolize foreign molecules with a wide range of chemical diversity. Instead of developing a tailored nucleophile to react with specific drug, general mechanisms are used to oxidize generic scaffolds found in all drug molecules. This greatly simplifies the extent of biological tools needed and improves genetic efficiency for drug metabolism while maintaining a broad reactivity and substrate scope.
Furthermore, not every single oxidation, reduction, hydration, etc., that can occur will occur. The above examples outline potential reactions with sp3 and sp2 carbons, heteroatoms, acids, and bases. Many drugs contain multiple functional groups, and there is usually a preference for specific metabolite profiles that depends on multiple features including the steric hinderance, electrophilicity, and polarity of the molecule. Once the molecule undergoes an initial modification, it may be sufficiently water-soluble to be expelled without further modification. However, in the same way, a single modification may not be sufficient and there can be multiple modifications that occur.
A key part of medicinal chemistry programs is identifying the metabolic liabilities on the drug – positions that are metabolized. This can help extend the lifetime of the drug or identify Phase I and Phase II metabolites. A metabolite identification (MetID) experiment is performed where a compound is subjected to hepatocytes and the resulting products are identified through mass spectrometry. The resulting pathway may not be linear and may demonstrate different transformations occurring at the different stages although they all converge on the common goal of preparing the compound for elimination.
