2 How are Drugs Discovered?
“The best way to discover a new drug is to start with an old one.”
– James Black
2.1 Natural Products and Natural Product Analogs
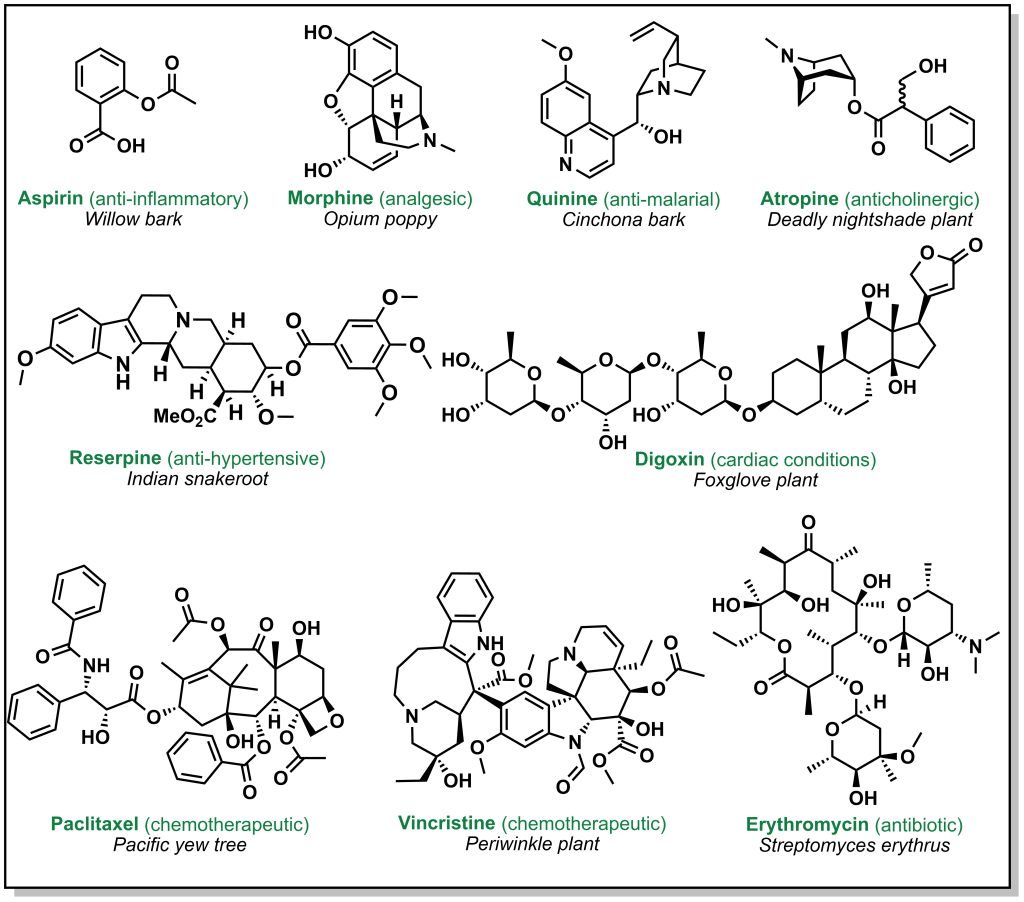
Drug discovery is not always carried out at a molecular level. Herbs, berries, roots, barks, and other natural products have been go-to ailments for humans since antiquity. These natural medicines were often ‘prescribed’ without any prior knowledge of their active constituents. Over time, multiple ancient populations accumulated their own pharmacopeia for natural products, several of which have served as inspiration for the isolation and/or synthesis of more innovative and potent compounds. These remedies derived from plants, animals, or microorganisms have foundational properties that transformed the landscape of modern medicine. Figure 2.1 highlights currently employed drugs that were derived from natural sources along with a selection of case studies below.
2.1.1 Aspirin
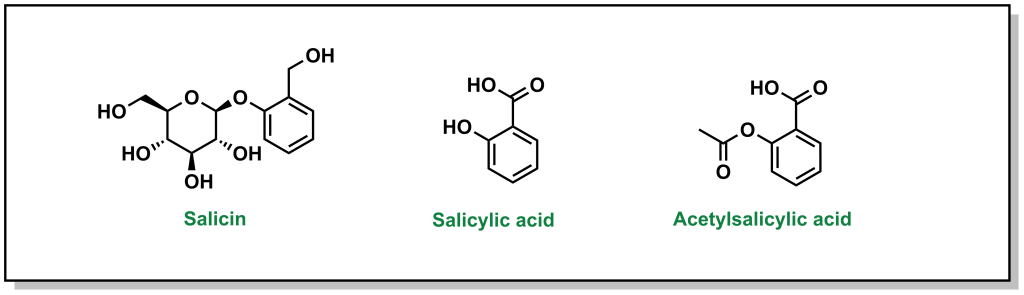
The discovery of aspirin and its growth to one of the most widely used over-the-counter analgesic (“pain-relieving”) drugs, spans multiple millennia, and involves many landmark developments in isolation and synthetic chemistry. Different civilizations independently converged on the use of willow trees, particularly the white willow (Salix alba) and meadowsweet (Filipendula ulmaria), for fever-reducing and pain-relieving properties. The earliest documented use can be found in the Ebers Papyrus, an ancient Egyptian medical text (3000 BC) although Reverend Edward Stone (1763) is often credited with the first scientific study in demonstrating the therapeutic properties . In 1828, Professor Joseph Buncher (Germany) was able to extract the active ingredient (which he named salicin), and it was later refined to salicylic acid by French chemist Charles Gerhardt (1853). Dr. Felix Hoffman (a German chemist at Bayer) was able to acetylate the compound, which alleviated several adverse side effects associated with salicin and salicylic acid, resulting in the compound acetylsalicylic acid (Figure 2.2). This landmark discovery represents the first synthetic drug from pharma, which galvanized a new era in therapeutics. Bayer registered the drug under the name Aspirin in 1899, with the namesake as a derivatization from yet another natural species of meadowsweet (Spirea ulmaria).
2.1.2 Morphine and Codeine
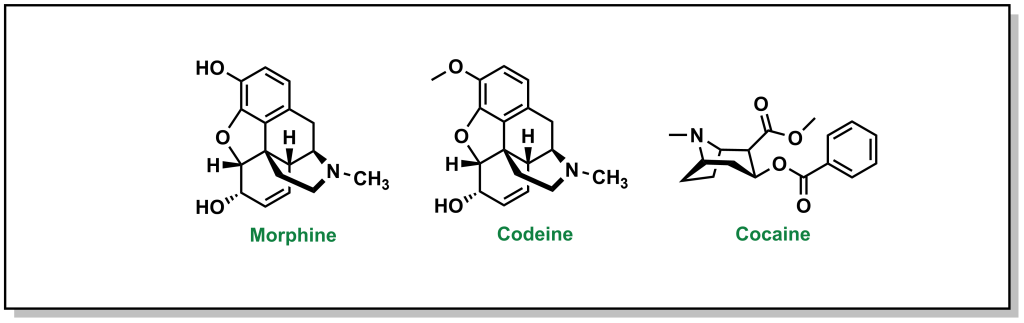
The powerful analgesic properties of the poppy plant (Papaver somniferum) were also known by multiple civilizations, with archaeological evidence of human use as far back as 5000 BC in the Mediterranean region. The seed capsules of poppy plants contain a milky substance called latex that contains opium. Opium is comprised of a mixture of chemical compounds called alkaloids (a loosely defined subset of naturally occurring organic compounds containing at least one nitrogen). In 1804, the German pharmacist, Friedrich Sertürner, isolated morphine (Figure 2.3) from opium (with the name emerging from Morpheus, the Greek god of dreams). Similarly, French chemist Pierre-Jean Robiquet was able to isolate codeine (parent compound of morphine) as another component from opium in 1832. Codeine was named after the Greek word “kodeia” referring to poppy head. Other euphoria-inducing drugs can also be synthesized from opium including heroin and oxycodone, although these are all tightly controlled substances due to their heavily addictive properties.
2.1.3 Penicillin
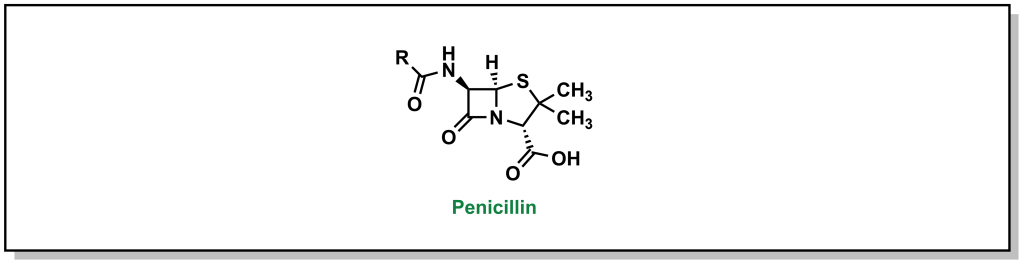
In 1928, Scottish physician, Alexander Fleming, was conducting research on the growth properties of the bacteria, Staphylococcus aureus. He returned from a two-week vacation and discovered that his cultures were contaminated by mold, but also that the bacteria could not grow in close proximity to the fungi on the culture plate. The fungi was determined to be Penicillium notatum. After culturing it, Fleming confirmed that the growing broth harboured an anti-bacterial substance that was effective on Gram-positive pathogens but not on Gram-negative bacteria. In 1929, he re-named the broth, penicillin, in place of the more colloquial name, “mold juice”. The chemical structure was determined and purified by another team (Howard Florey and Edward Chain) in 1940, followed by the first treatment in patients in 1942. (Figure 2.4) Its uptake into the clinics was largely accelerated during World War II with its success in tackling bacterial infections and the trio of scientists awarded the Nobel Prize in Physiology or Medicine in 1945.
2.2 Modern Rational Drug Design
One of the common themes evident in drug discovery via natural products is that it is grounded in astute observation of often-serendipitous circumstances, followed by empirical trial-and-error methods, and ultimately isolation of the key active ingredient. Over the early 20th century, this framework for drug discovery transitioned to a more step-wise hypothesis-driven approach with the goal of systematically developing, synthesizing, and exploring new (non-natural) composition-of-matter. This reform in therapeutic methodology was accelerated by the creation of the US FDA in 1938, followed by substantial administrative changes in the Kefauver-Harris Drug Amendments of 1962, as a result of the thalidomide drug scandals. The US FDA oversees public health in the US including the approval of new drugs and medical devices. The Canadian counterpart is referred to as Health Canada, which was instituted in 1919 in response to the Spanish Flu outbreak, and later reformed in 1993. Both Health Canada and the US FDA collaborate closely (via the Regulatory Cooperation Council Joint Forward Plan) to harmonize several drug-related policies, although they are separate entities and can/will uphold different guidelines.
2.3 Typical Stages of the Drug Discovery Pipeline
For a medicinal chemist, the stages of the drug discovery pipeline are often represented in a linear, forward-moving strategy, although the overall process is highly iterative and can involve multiple cycles of optimization and re-visiting of each stage (Figure 2.5).

2.3.1 Stage 1: Target Selection
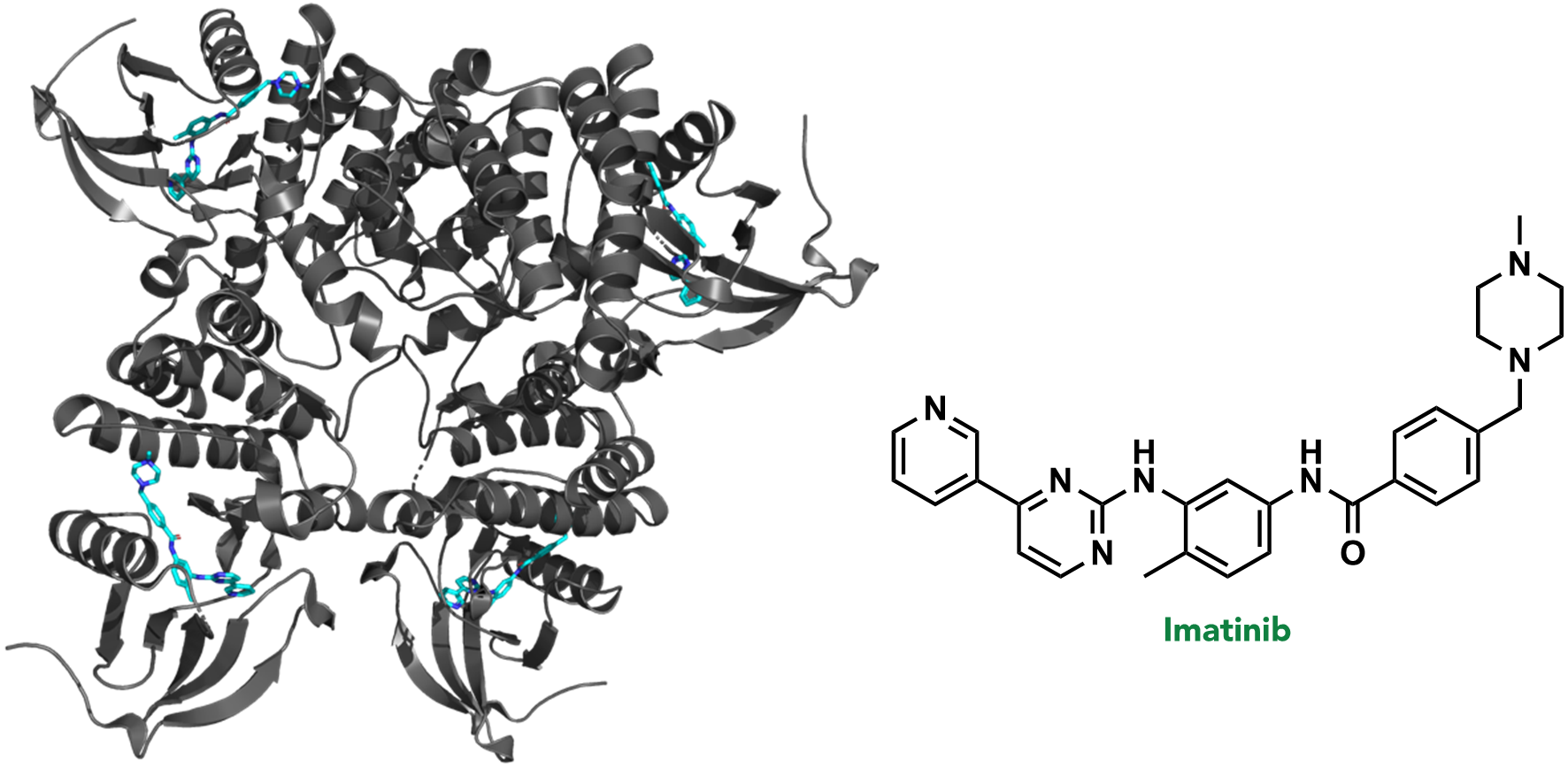
In the first stage of drug discovery, a target or specific disease is selected. A target refers to a biomolecule where pharmacological or genetic intervention would (directly or indirectly) disrupt a critical pathological biochemical process. For example, >95% of chronic myeloid leukemia (CML) cases result from a random genetic mutation that creates the fusion protein, BCR-ABL kinase. BCR-ABL kinase is therefore a target in CML, and is often treated with the drug, imatinib (or one of its analogues). (Figure 2.6)
One disease can have more than one target, and similarly, one target may be relevant in more than one disease. The rationale for selecting a target is usually supported by an array of genetic, biological, and physiological data that suggest it will be efficacious to knock-down while not compromising overall safety to the patient. This is generally referred to as target validation. For example, disrupting the enzyme Protein Kinase A (PKA) is not a feasible option in the rare fibrolamellar hepatocellular (FLC), despite the fact that its variants have been shown to drive this cancer. This is because of the importance of PKA in the heart for maintaining appropriate cardiac functioning. However, there are many cases where background biological information is not readily available, or the target is not well-studied. In these cases, a greater importance is placed on pre-clinical investigations and safety trials.
2.3.2 Stage 2: Hit Identification and Hit-to-Lead / Lead Discovery
Following selection of the disease/target, the next stage involves identifying a chemical compound that can interact with the target. Usually, a series of compounds are evaluated in an experiment that involves the desired target. This experiment, called an assay, can involve a variety of tools to quantify the interaction between the compound and biomolecular target; for example, monitoring the interaction (binding event) via nuclear magnetic resonance (NMR), quantifying a change in the target activity via fluorescence, or observing cell death via imaging. The types of assays will vary, depending on both the target and the nature/origin of the compounds being tested, and are often custom-designed for each drug discovery program.
The output of the assay will usually provide an initial group of compounds that can engage with the target, which are referred to as hit compounds. The activity of these hit compounds can vary substantially, but they serve as general starting points to create additional generations of molecules with better activity. The goal of subsequent organic synthesis campaigns is to improve the hit compounds and create/select a lead compound. A lead compound is a compound that:
- Interacts with the target to achieve the desired biological activity.
- Is amenable to synthetic modifications.
- Can reach the target once administered.
Distinguishing and selecting lead compounds will be discussed later in this text. However, an initial hit molecule differs from a final lead molecule in that it usually has lower biological activity, and has not been validated to reach the target upon administration. As such, these two properties are classical outcomes of hit-to-lead or lead discovery campaigns, where the goal is to chemically synthesize new molecules that build and optimize a hit compound into a lead molecule.
2.3.3 Stage 3: Lead Optimization
Although identifying a lead molecule is a monumental step forward in the drug discovery pipeline, it is often chemically distant from the final drug candidate. This is usually because the lead molecule has specific properties that can limit its efficacy. For example, the structure-activity relationship (SAR) optimization rounds from the previous hit-to-lead studies may have identified specific functional groups required for target binding (biological activity), but the oral absorption of the compound may not be sufficient, as a result of these chemical groups. Therefore, at the lead optimization stage, the goal is often to preserve the structural elements that maintain potency of the lead compound, while optimizing a new molecule with improved physical properties for bioavailability. It is conceivable that potency of the newly developed candidate molecules may be reduced, as other parameters are optimized. This is a balancing act to fine-tune the optimal overall properties between potency, efficacy, permeability, and stability.
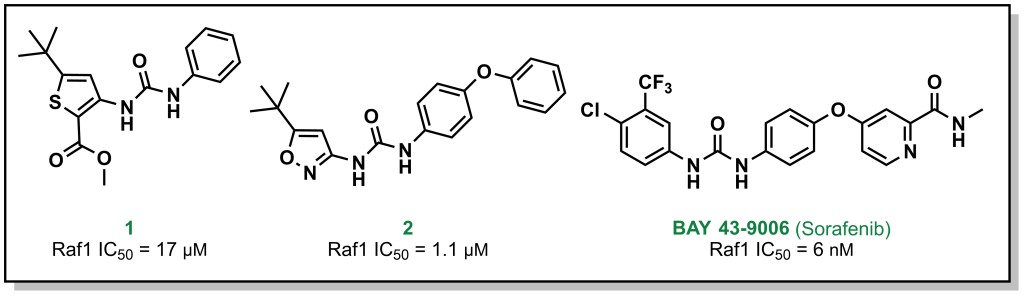
For example, Bayer (in partnership with Onyx Pharmaceuticals) initially screened ~200,000 compounds to explore the target Raf1 kinase, which led to the initial hit molecule 1 (Figure 2.7) with a half-maximal inhibitory concentration (IC50) of 17 µM. The urea and phenyl group were identified to be critical for potency and another ~1000 bis-aryl urea analogs were generated that led to an isoxazole derivative as the lead compound (2) (Figure 2.7). The compound underwent lead optimization to the final development candidate BAY 43-9006 (sorafenib) with a potent affinity for Raf-1 (IC50 = 6 nM). Crystallographic studies (which were determined retrospectively) revealed that the urea moiety was crucial in the SAR, due to the formation of two critical H-bonds (with the backbone aspartic acid residue of the DFG loop and the glutamate side chain of the αC helix of the target protein – structural elements that will be discussed in Section 2.5.1). Additional key features from BAY-43-9006 that were identified include the 4-pyridyl ring that mimics the natural adenine scaffold and the chlorobenzene ring which interacts with a hydrophobic pocket behind the orthosteric ATP binding site. Although the lack of structural information did not allow for visualization of these effects at the time of compound synthesis, these features improved the binding capacity of the drug which was represented by the lower IC50 values and indicated that they were important in the lead optimization process.
2.3.4 Stage 4: Preclinical / Clinical Development & Regulatory Approval
The focus of this resource is on medicinal chemistry, and following completion of lead optimization and pre-clinical trials, the compound is generally optimized and there is a diminished motivation for synthesizing newer compounds. However, the subsequent regulatory steps are briefly summarized below.
Following lead optimization, the top developmental candidate needs to be evaluated in more complex models. This usually involves a mammalian model such as a mouse or rat that has been modified to express a phenotype representing the disease state. For example, exploring cancer tumors can involve engrafting specific cancer cells into a mouse and monitoring the tumor volume upon compound treatment (as a measure of cancer-killing potential). For neurological diseases, this could involve genetically introducing known mutations and monitoring specific exercises such as novel object recognition time tests (as a measure of memory function). Generally, these studies are benchmarked to the standard-of-care which is the current drug regimen used in clinical settings, and this provides an understanding of the degree to which the candidate molecule can improve outcomes. Although it is not an absolute requirement, usually two preclinical models are required to advance to human trials.
Clinical trials are an expensive and complex endeavour with the goal of demonstrating that the drug candidate is safe and efficacious. Biotech companies looking to evaluate their drug candidates in humans (in Canada) require approval from Health Canada. The classical route involves Phase I trials, which are predominantly focussed on safety and pharmacokinetics of the drug candidate in healthy individuals. However, for rare diseases, the compound may be administered to a population with the disease of interest in Phase I. Following positive results, a Phase II clinical trial would be initiated with a cohort of patients from the disease population. Phase III clinical trials would have an even larger disease population for improved sampling statistics. Depending on the type of dosing performed, the clinical trial may also be labelled with an a or b suffix. Clinical trials cost millions of dollars and require years to prepare clinical centres and teams, recruit patients,
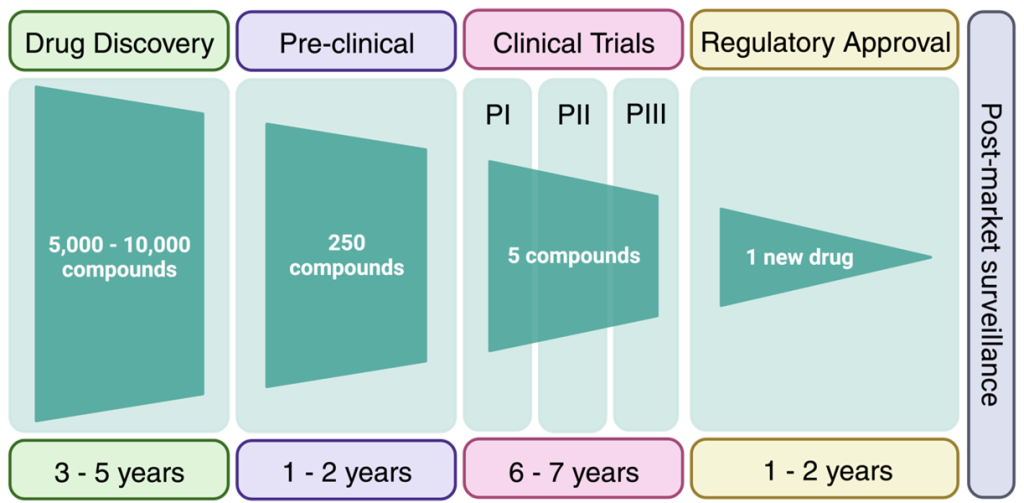
administer the drugs, and analyze the results. On top of this, the success rate of a drug to advance through all three clinical trials with positive data is ~7%. Overall, it is estimated that advancing a compound through the drug discovery pipeline may take 15-20 years and cost 1-2 billion dollars. Although this may seem daunting, understanding why drugs fail at the late stage, helps front-load these issues into the medicinal chemistry thought process at the beginning of the drug discovery pipeline. The results of many clinical case-studies have revealed benefits and challenges of different functional groups and the goal of medicinal chemistry is to build on these learnings to avoid attrition of drugs at late clinical stages.
2.4 Rational Drug Design: The “Magic Bullet” Concept
Paul Ehrlich, a German physician, pioneered a number of concepts that took a foothold in the early applications of medicinal chemistry. One of his primary contributions was in the field of immunity, for which he was awarded the Nobel Prize in Physiology or Medicine in 1908. He developed a “side-chain theory” which purported that specific chemical structures could elicit a response by immune cells in the blood, similar to the protein-ligand “lock-and-key” biochemistry model. He also adopted the concept of a “magic bullet” (a concept from German stories of a bullet that locks on to a target and cannot miss once fired), proposing that it would be possible to develop a compound that mimics these properties and specifically engage invading foreign entities with a high degree of specificity.
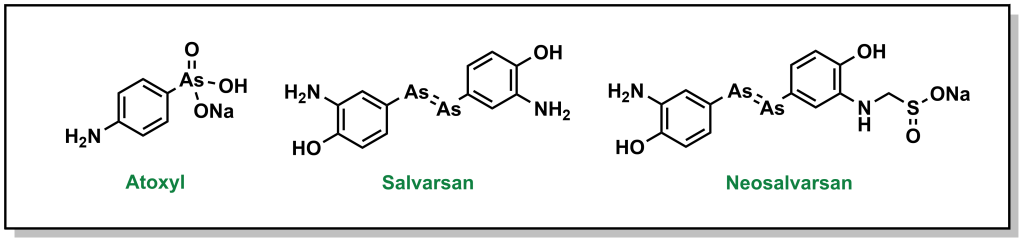
Ehrlich pursued his idea of a magic bullet, generating libraries of compounds based on the toxic drug atoxyl for the treatment of syphilis, which was a major public health threat in Europe at the time. His goal was to identify a compound that would selectively kill the responsible bacterium, Treponema pallidum, without harming healthy cells. Eventually, his team developed compound 606 or arsphenamine which was marketed to Hoechst AG under the name salvarsan in 1910. Although this compound was a substantial improvement over the standard-of-care (mercury-containing compounds), it was highly unstable in air, which could lead to multiple and serious adverse side effects. Another 300 derivatives later, his team developed a more degradation-resistant drug, which was called neosalvarsan (Figure 2.8). Although these compounds were superseded by the discovery of penicillin in the 1940s, the approach adopted by Ehrlich foreshadowed the iterative drug discovery pipeline to optimize new selective compounds.
2.5 Rational Drug Design: Understanding the Target
Understanding the target will shape the drug discovery program and alter the overall medicinal chemistry strategy. If considering just the human genome, there are approximately 20,000 genes that encode potential targets. About 15% of the genome can be pharmacologically altered with today’s collection of drugs. This means that there are potentially thousands of targets without drugs (although not all of these proteins are disease-relevant or would be significant effectors of disease if targeted by a drug). Additionally, there are a wealth of non-human proteins, such as pathogenic biomolecules from bacteria or viruses that cannot be accessed by current drugs.
Although there are many different pathways, and therefore many protein targets that require pharmacologic intervention, current targets are asymmetrically skewed toward certain privileged protein families. For example, nearly two-thirds of all drugs on the market, target proteins from either the kinase or G-protein-coupled receptor (GPCR) superfamilies. Part of the reason why these protein classes are over-represented in drug discovery is because they exist at the top of large biochemical cascades, and therefore blockading their action can yield powerful responses. GPCRs and nuclear receptors have many different types of drugs that act on the same target (i.e. there are many selective drugs for these targets), whereas for kinases the reverse trend is true – there are many different kinases that are targeted by a smaller number of drugs, which indicates many more “off-targets”. Additionally, GPCRs are located on the cell surface, which reduces the requirement of a drug to cross an additional membrane. Importantly, kinases have been heavily studied both in structure and function which helps guide the drug discovery process.
Designing drugs based on protein structure represents a critical step in the rational development of drugs based on protein-ligand interactions. Although the “lock-and-key” model of protein-ligand binding has been updated to involve “induced-fit” models that account for the fluidity of the protein and ligand conformations, it still provides a useful classical analogy for understanding the goals of designing new drugs for protein targets. This model has been applied to understand the different conformations of kinases and how drugs can engage these targets.
2.5.1 Kinases
Protein kinases are enzymes that are responsible for transferring a phosphate group from ATP to another protein with a hydroxy-side chain (e.g. tyrosine). This post-translational modification introduces a di-anionic charge which is unique to a phosphorylated residue (and not found on any naturally incorporated amino acid). As such the consequences of phosphorylation can result in drastically altered protein conformations or interaction partners. There are two main types of kinases, Serine/Threonine kinases (~385 proteins) and Tyrosine Kinases (90 proteins) which specifically phosphorylate their namesake residue. Phosphorylation via Ser/Thr kinases is usually associated with large-scale conformational changes whereas as action by Tyr kinases can result in protein localization changes. There are also rarer kinases that are capable of phosphorylating non-conventional residues such as Histidine, Arginine, and Lysine.
The kinase structure is highly conserved and the secondary structural elements are well-defined. The active kinase domain contains two lobes, a small N-lobe (largely comprised anti-parallel β-sheets) and a large C-lobe (largely α-helical) connected by a hinge region. The ATP binding site is sandwiched at the interface of these lobes and the adenine ring forms H-bonds with the hinge region.
Kinases cycle between an inactive (open) conformation and an active (closed) conformation. The open conformation enables ATP / ADP to access the active site hinge region, whereas the closed conformation facilitates formation of the functionally active cleft. Both the N- and C- lobes have key structural features associated with each state. The N-lobe contains a large helix (labelled an αC-helix) that forms the roof of the active site and will either rotate away (open/inactive) or towards (closed/active) the orthosteric site. The C-lobe contains a conserved loop (referred to as an Activation or DFG loop) that begins with residues Asp, Phe, and Gly and ends with Ala, Pro, and Glu. The Asp side chains bind Mg2+ which helps coordinate the phosphate of ATP. In the active conformation, the Asp faces into the pocket, whereas the inactive conformation has the Asp extruding out of the pocket. (These conformational switches also have the Phe entering and blocking the adenine binding pocket).
Kinases represent the archetypical drug binding sites – they contain a well-defined binding pocket that is buried, hydrophobic, with affinity for a known ligand (ATP). In fact, the binding pockets of kinases and the inhibitors that have emerged are so extensively studied that they are stratified across 6 types:
- Type I inhibitors bind directly at the orthosteric (ATP binding) site and leverage conserved catalytic residues in their interactions. These inhibitors are ATP-competitive and have excellent shape complementary by exploiting the rigid nature of the active site. For example, crizotinib and dasatinib are Type I kinase inhibitors.
- Type II inhibitors bind at the ATP binding site but in the DFG-out (inactive) conformation. In this conformation, a new hydrophobic site is exposed between the C-helix of the small lobe and the DFG motif. The example of sorafentib, which is discussed above, is a Type II inhibitor.
- Type III inhibitors bind deeper in the ATP binding site, in a hydrophobic pocket behind the active site. These inhibitors have substantially improved selectivity, since this hydrophobic pocket is kinase-specific. Moreover, these inhibitors do not have a heterocyclic group that normally mimics the adenine ring. However, this pocket is often transient and occupancy does not guarantee inhibition.
- Type IV inhibitors bind at an allosteric site, completely distinct from the ATP site.
- Type V inhibitors are bivalent inhibitors with chemical moieties that engage with structural elements on both lobes of the kinase.
- Type VI inhibitors are covalent inhibitors that will form a covalent bond with a residue (usually cysteine) on the kinase.
An important aspect for inhibitors is that over time, cells can become resistant to the effects of inhibitors. This can emerge from mutations in the target that reduce binding affinity or capacity of the inhibitor. In kinases, this can often occur with a gatekeeper residue, which is a key residue that is located close to the hinge region and guards access to the pockets behind the adenine ring. In BCR-ABL1 (CML) the T315I gatekeeper mutation results in steric hindrance that impedes binding of imatinib. In EGRR the T790M gatekeeper mutation induces resistance by increasing affinity for ATP. New drugs need to be designed that can overcome these mutations by avoiding the gatekeeper, allosterically engaging the target, or harnessing a separate biochemical pathway altogether. For example, the drug ponatinib bypasses the gatekeeper mutation via an ethynyl group, whose linear structure sterically evades the bulky T315I mutation and can continue to block BCR-ABL activity.
