30 2.3: Amyloid-β Role in Alzheimer’s Disease
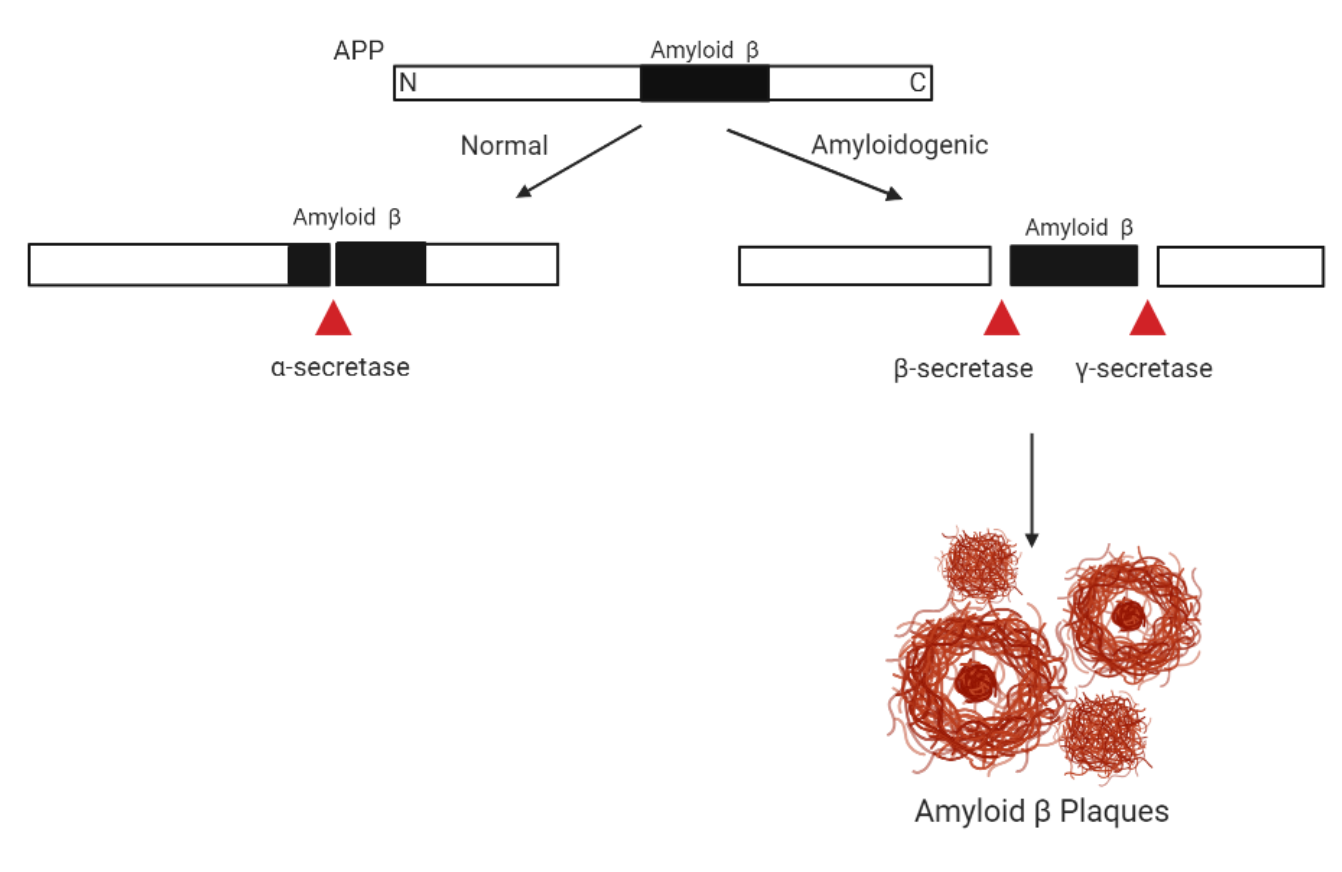
In 1992, Hardy and Higgins presented The Amyloid Cascade Hypothesis that “Our hypothesis is that deposition of amyloid-β protein (AβP), the main component of the plaques, is the causative agent of Alzheimer’s pathology and that the neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition” (Hardy and Higgins). Amyloid-β is originated from a large protein called amyloid precursor protein (APP) which is cleaved to two peptides by proteases called β- and γ-secretases producing Aβ(1-42) peptide and Aβ(1-40) peptide (Murphy and LeVine). Even though there is a more abundant amount of Aβ(1-40) found in the brain, Aβ(1-42) is found to be more neurotoxic and more readily to aggregate. It has been shown that elevated level of Aβ(1-42) is associated with the pathogenesis of Alzheimer’s disease. This is because soluble Aβ(1-42) can bind to each other to form oligomers and aggregate which form to insoluble Amyloid-β plaques (Brothers et al.). Ultimately, aggregation disrupts cell-to-cell communication and activates immune cells which then cause inflammation.
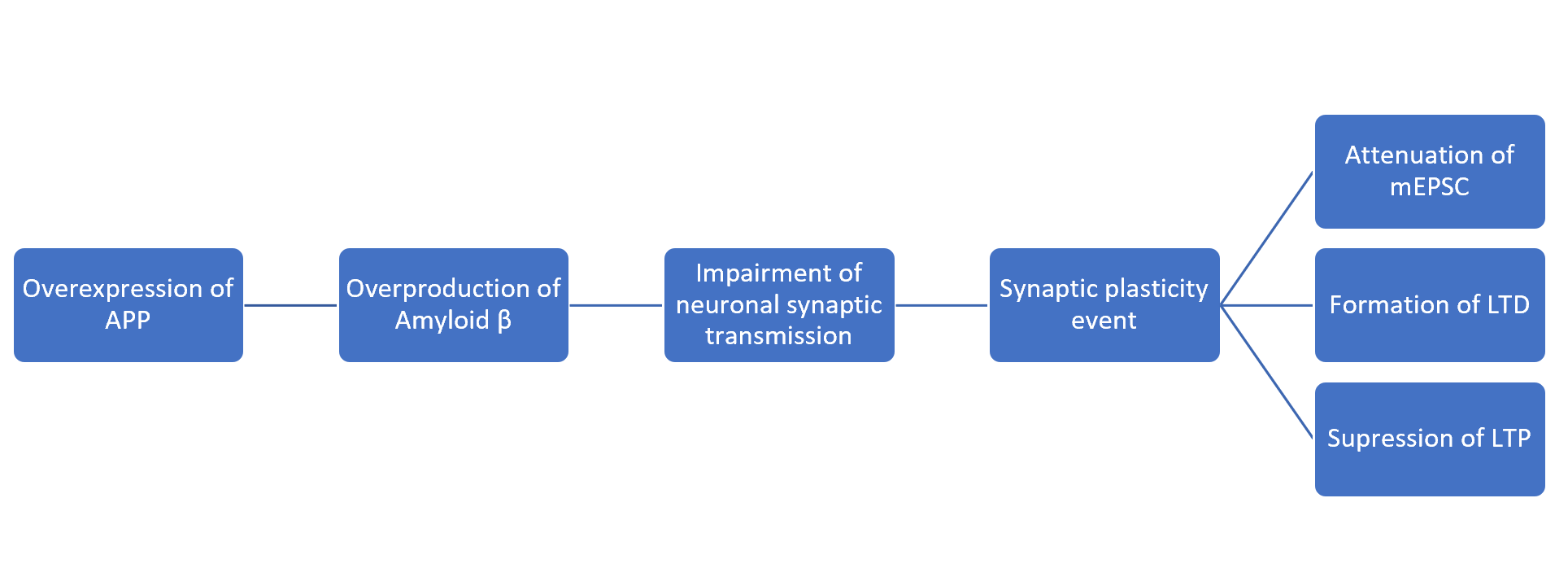
From previous mice studies, it has been shown that overexpression of APP results in the reduction in the excitatory neuronal synaptic transmission both presynaptic and postsynaptic. This event then results in the impairment of miniature excitatory postsynaptic currents (mEPSC) (Ting et al.). Additionally, another study has shown that APP knockout mice were underweight and locomotor activity which suggests the importance of this protein (Zheng et al.).