5 Traitements médicamenteux de la douleur et de l’inflammation
5.1 Pathologie
Une lésion tissulaire suscite généralement une réaction physiologique et psychologique sous la forme d’inflammation et de douleur. Bien que ces réactions soient souvent perçues négativement, l’inflammation aiguë et la douleur jouent un rôle crucial dans l’élimination des stimuli négatifs, la lutte contre les envahisseurs étrangers et le processus de guérison des blessures. Toutefois, l’inflammation et la douleur prolongées peuvent provoquer encore plus de dommages et d’inconfort. De nombreux médicaments ont été développés pour soulager la douleur et l’inflammation en ciblant les réponses physiologiques ou psychologiques.
Prenons un exemple simple : lorsqu’une lésion cellulaire survient, causée par une perforation ou une rupture de la membrane, elle déclenche une cascade de réponses variées. Un premier aspect à considérer est l’afflux de Ca2+ dans la cellule. Le Ca2+ joue un rôle essentiel comme messager biochimique puissant. De nombreuses protéines et facteurs de transcription sont sensibles à sa liaison, ce qui peut déclencher diverses réponses selon le type cellulaire. L’une de ces réponses principales est l’activation d’une protéine appelée phospholipase A2 (PLA2). La PLA2 interagit avec les phospholipides de la bicouche cellulaire et clive les acides gras des phospholipides.
{kind=link}
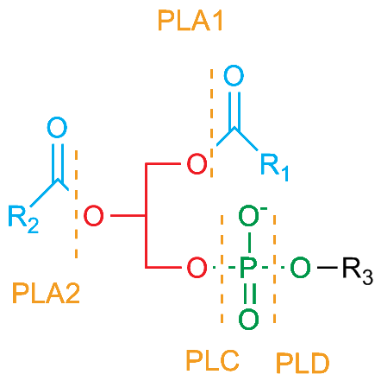
Rappelons que la structure d’un phospholipide comporte une structure de base de glycérol avec une tête de phospholipide estérifiée sur l’un des atomes d’oxygène du glycérol (appelé selon la numérotation stéréospécifique 1 ou SN1), ainsi que des chaînes d’acides gras estérifiées sur les deux autres atomes d’oxygène (désignés comme SN2 et SN3) (Figure 5.1). Les différentes enzymes phospholipases clivent chacune à des positions SN précises : PLA1 clive à la position SN1, PLA-2 clive à la position SN2, PLAB clive à la position SN1 ou SN2 et PLAC ainsi que PLCD clivent sur des côtés spécifiques du phosphate à la position SN3. Lors de la réponse inflammatoire, l’une des phospholipases les plus importantes est la PLA2, qui clive au centre de l’acide gras à la position médiane (SN2). L’acide gras libéré est souvent un acide arachidonique (Figure 5.2).
5.1.1 L’acide arachidonique et l’inflammation
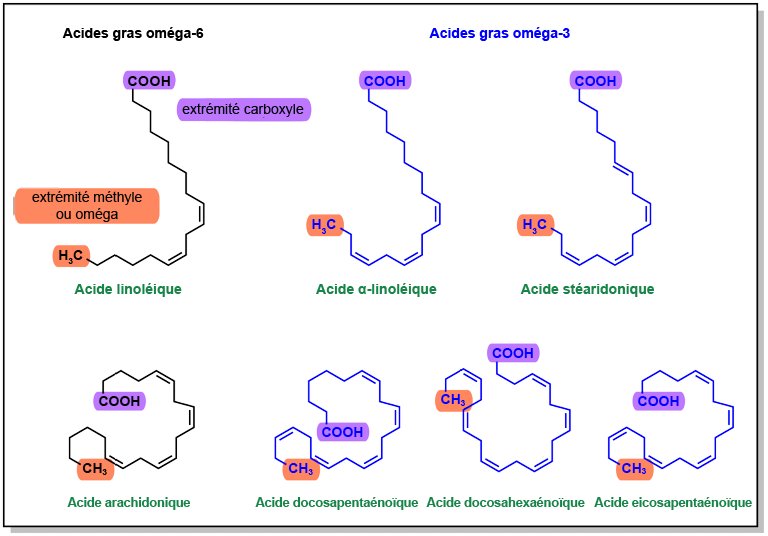
Plusieurs types d’acides gras sont incorporés dans les phospholipides. Les acides gras physiologiques variants par leur longueur et leur degré de saturation, il a toujours été plus pratique de les nommer à partir de l’extrémité oméga (côté carbone terminal) plutôt qu’à partir de l’extrémité alpha. Les acides gras oméga-3 possèdent une double liaison au niveau du C3 à partir de la position oméga, tandis que les acides gras oméga-6 ont une double liaison au niveau du C6 à partir de la position oméga (Figure 5.2). L’acide arachidonique, un acide gras oméga-6, possède quatre doubles liaisons. Ces liaisons sont orientées en configuration « cis », ce qui confère à l’acide arachidonique une géométrie plus refermée sur elle-même qui contribue à maintenir la fluidité de la membrane cellulaire. Ses doubles liaisons sont particulièrement sujettes à l’oxydation, ce qui fait de cet acide gras une cible privilégiée pour les voies de signalisation en aval (Figure 5.3).
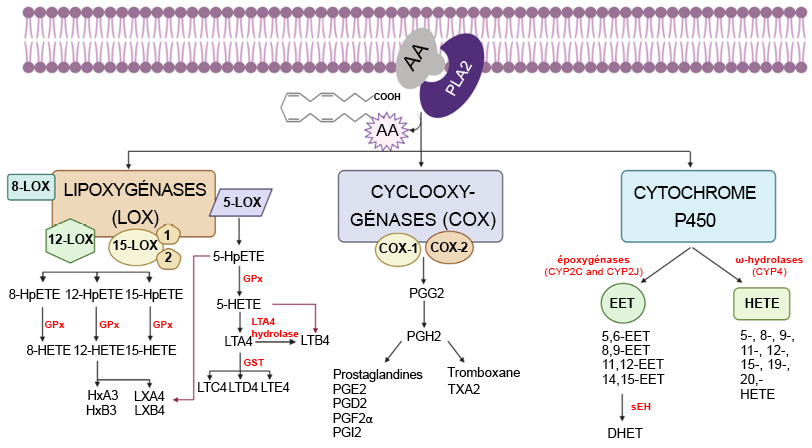
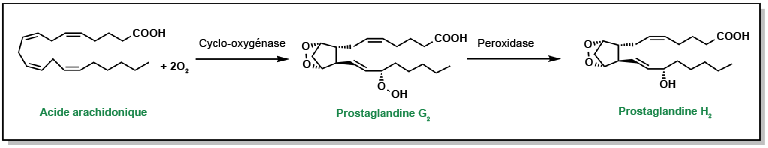
Les voies de signalisation de l’acide arachidonique sont amorcées suite à l’afflux de Ca2+ et à l’activation de la PLA2. Cette dernière clive des phospholipides spécifiques, libérant ainsi l’acide arachidonique de la position SN2. L’acide arachidonique libre joue le rôle de substrat pour diverses protéines. Ces protéines se répartissent principalement en trois classes : les cyclooxygénases (enzymes COX), les enzymes du cytochrome P450 (CYP) et les lipoxygénases (enzymes LO). L’acide arachidonique est oxydé en différentes molécules appelées eicosanoïdes, comprenant des molécules telles que les prostaglandines et les thromboxanes. Chacune de ces molécules peut engendrer différents effets, tels qu’une inflammation accrue ou l’agrégation des plaquettes.
L’inhibition de cette voie peut se faire à divers niveaux. La PLA2, se trouvant au tout début de la voie, est fortement inhibée par les stéroïdes, qui interrompent la réponse inflammatoire. Cependant, les stéroïdes sont des hormones et peuvent avoir des effets multiples sur l’ensemble du corps. Par conséquent, les enzymes situés à la base de la voie de réponse biochimique, tels que les métaboliseurs de l’acide arachidonique, constituent une stratégie mieux ciblée. L’approche la plus courante consiste à inhiber les enzymes COX responsables de la production de prostacyclines, de prostaglandines ou de thromboxanes
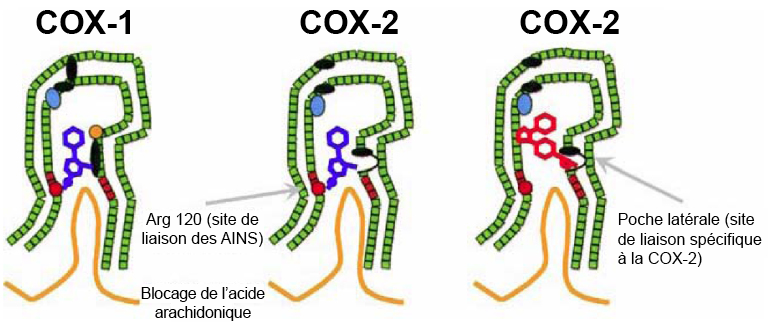
Il existe deux isoformes principales de ces enzymes, appelées COX1 et COX2. La COX1 est active de manière constitutive, bien qu’à de faibles niveaux dans de nombreux tissus, et contribue au maintien de l’homéostasie. La COX1 joue un rôle essentiel dans la préservation de la paroi de l’estomac, le fonctionnement normal des reins et l’agrégation des plaquettes. La COX2 est surexprimée en réponse aux signaux inflammatoires. Lors de leur activation, les enzymes COX1 et COX2 se localisent à la surface de la membrane cellulaire. Elles se positionnent de manière à ce qu’un long canal à l’intérieur de l’enzyme soit orienté vers la membrane, permettant ainsi d’accueillir les molécules d’acide arachidonique libres (Figure 5.4).
Lorsque l’acide arachidonique se lie aux enzymes COX, il subit une oxydation pour former la prostaglandine G2 qui est ensuite convertie en prostaglandine H2, cette dernière agissant comme substrat pour d’autres enzymes (Figure 5.5).
5.2 Traitements de la douleur et de l’inflammation
Étant donné que les molécules qui ciblent les enzymes COX ne sont pas des stéroïdes, mais qu’elles réduisent néanmoins l’inflammation, celles-ci sont appelées anti-inflammatoires non stéroïdiens ou AINS. Bien que l’enzyme COX ait été isolée et purifiée pour la première fois en 1976, elle était à ce moment inhibée par l’action de produits naturels utilisés par de nombreuses civilisations, tels que l’écorce du saule blanc ou de la reine-des-prés. L’ingrédient actif de ces plantes, l’acide salicylique, a finalement été identifié. Pour réduire les effets indésirables, comme l’inconfort gastro-intestinal, l’acide acétylsalicylique estérifié (plus connu sous le nom d’aspirine) a été développé, breveté et commercialisé en 1899. L’aspirine représente une étape importante dans l’histoire des agents pharmaceutiques.
5.2.1 Aspirine
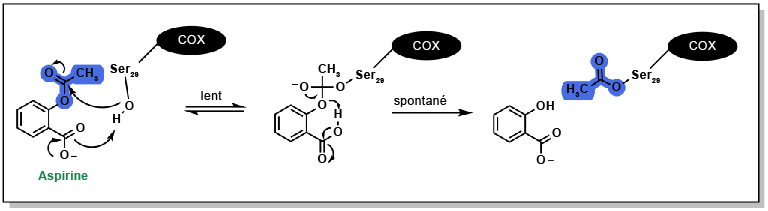
Les effets anti-inflammatoires de l’aspirine résultent de l’inhibition de l’enzyme COX. L’acide acétylsalicylique se lie à la chaîne latérale chargée positivement de l’Arg120 sur l’enzyme COX et empêche l’acide arachidonique de pénétrer dans le canal protéique, où il serait oxydé en métabolites inflammatoires (Figure 5.4). L’aspirine possède aussi un mécanisme d’action unique : elle forme une liaison covalente (ou irréversible) avec l’enzyme COX en réagissant avec la Ser29 (Figure 5.6). Le groupe acétyle agit comme un groupement partant, et l’enzyme COX est modifiée de manière permanente par l’acide salicylique. Pour rétablir leur fonction, les cellules (comme celles de la muqueuse gastrique) doivent biosynthétiser de nouvelles enzymes COX1/COX2. Cependant, les cellules non nucléées (telles que les plaquettes) ne peuvent pas synthétiser à nouveau ces protéines, ce qui fait de l’aspirine un puissant inhibiteur de l’agrégation plaquettaire.
5.2.2 Autres médicaments anti-inflammatoires non stéroïdiens
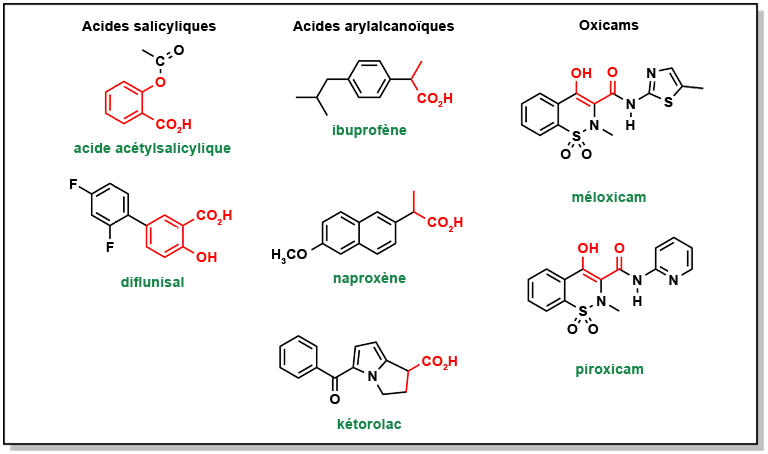
Il existe plusieurs autres AINS, en dehors de l’aspirine, mais ils se distinguent par un mécanisme d’action non covalent. On peut généralement les classer en trois catégories : les acides salicyliques, les acides arylalcanoïques et les oxicams (Figure 5.7). Dans tous les cas, ils possèdent une fonctionnalité acide qui est essentielle pour interagir avec l’Arg120 de l’enzyme COX et bloquer stériquement l’accès à la poche active.
5.2.3 Tylenol
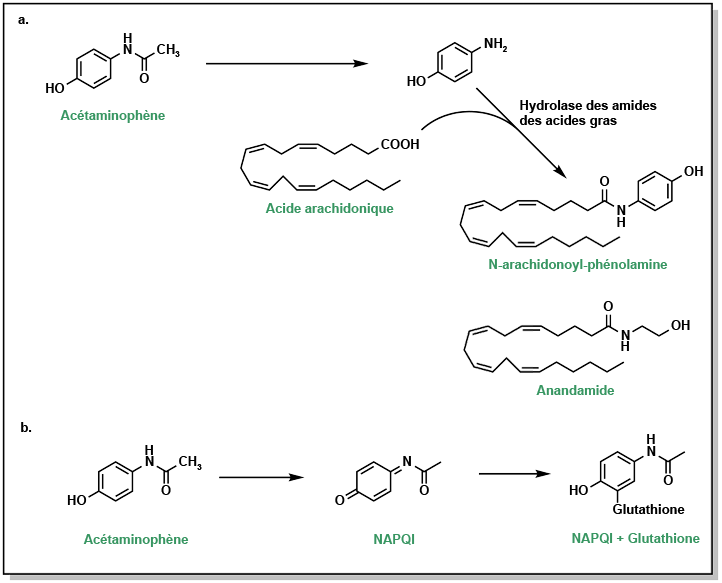
Le Tylenol, ou acétaminophène, est un analgésique couramment utilisé dont on croyait autrefois qu’il inhibait les enzymes COX. Cependant, des études ont montré qu’il se lie faiblement aux enzymes COX in vitro, ce qui le distingue des AINS, probablement en raison de la faible acidité du groupe hydroxyle. L’acétaminophène agirait au niveau central du cerveau pour bloquer les signaux de la douleur et interagir directement avec l’acide arachidonique (Figure 5.8a). Cependant, il est également capable d’induire des réponses anti-inflammatoires. Une des principales préoccupations liées au Tylenol est le risque de toxicité en cas de surdosage. Près de 95 % du Tylenol est métabolisé par glucurono et sulfoconjugaison (Figure 5.8b). Cependant, une petite quantité est métabolisée par le CYP2E1, ce qui conduit à la production de superoxyde et de NAPQI (N-acétyl-p-benzoquinone-imine) Cet intermédiaire est toxique, car il est très réactif et peut modifier de façon covalente différents résidus ou nucléotides. En présence de glutathion, la NAPQI est rapidement détoxifiée. Toutefois, si le Tylenol est consommé en grande quantité, le réservoir de glutathion hépatique s’épuise, laissant la NAPQI libre de réagir avec d’autres biomolécules.
5.2.4 Opioïdes
La douleur commence généralement au niveau des nocicepteurs dont le signal est transmis aux neurones secondaires du ganglion de la racine dorsale, puis au cerveau par l’intermédiaire des nerfs de la moelle épinière. La transmission du signal se fait au moyen de potentiels d’action, dont la fréquence augmente en fonction de l’intensité de la douleur. Les neurones libèrent de nombreux neurotransmetteurs qui favorisent différentes réponses :
- Glutamate : il active différents récepteurs sur le neurone post-synaptique (tels que NMDA, AMPA) et provoque l’entrée de Ca2+ et de Na+ dans la cellule, ce qui augmente sa charge positive.
- Substance P : elle entraîne la libération de substances telles que l’acide arachidonique et renforce le signal de la douleur.
- CGRP : il modifie l’expression du GPCR, ce qui renforce la réponse à la douleur.
Simultanément, l’organisme libère aussi des molécules endogènes qui atténuent certains effets de la douleur, comme les dynorphines et les endorphines. Ces molécules peuvent se lier aux récepteurs mu, delta ou kappa. Lorsque les récepteurs mu sont liés et activés dans le cerveau, ceux-ci produisent des effets euphoriques. Ces récepteurs diminuent la réponse des récepteurs GABA, ce qui entraîne une libération accrue de la dopamine (en éliminant les molécules qui limite sa libération). Ces ligands naturels produisent des effets analgésiques. Cependant, les opioïdes sont beaucoup plus puissants en tant que liants et produisent des effets analgésiques plus importants.
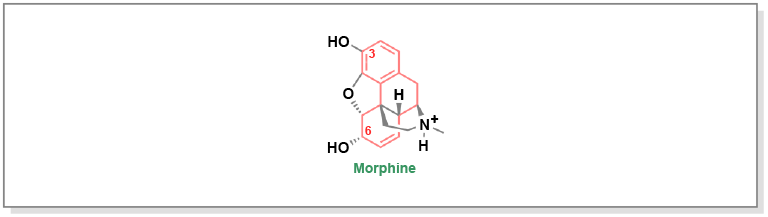
Les opioïdes, utilisés depuis des milliers d’années, proviennent du liquide laiteux (appelé latex) du pavot. La morphine, principal alcaloïde de la famille de l’opium, contient plusieurs groupes fonctionnels essentiels à l’activité analgésique (Figure 5.9). Ces groupes comprennent :
- l’azote tertiaire chargé positivement, qui s’engage dans la chaîne latérale d’un acide aspartique sur le récepteur mu
- l’azote tertiaire attaché à un carbone quaternaire par un pont double carbone
- le carbone quaternaire qui possède un groupe phényle attaché, favorisant les interactions d’empilement pi-pi avec le récepteur mu
- les groupes fonctionnels en C3 et C6 qui jouent un rôle crucial dans les interactions par liaison hydrogène
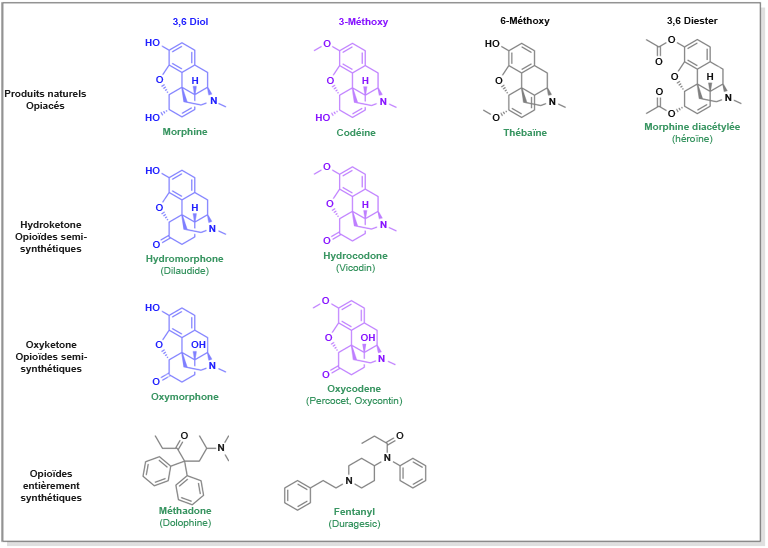
Les opioïdes, qu’ils soient naturels, synthétiques ou semi-synthétiques, ont fait l’objet d’études approfondies et présentent des propriétés alternatives en termes de biodisponibilité. Par exemple, la codéine, un opiacé naturel, diffère de la morphine uniquement par la présence d’un groupe méthoxy en position C3 au lieu de l’hydroxyle. Cette modification entraîne une augmentation significative de la lipophilie de la molécule. Cependant, le méthoxy en position C3 ne peut plus former de liaison hydrogène et son affinité avec le récepteur mu est nettement plus faible. La codéine pourrait se révéler extrêmement puissante si elle était métabolisée en morphine, mais cette transformation n’est pas efficace. En raison de la lenteur du métabolisme, bien que la lipophilie de la codéine soit supérieure à celle de la morphine, une dose orale de 100 mg est nécessaire pour obtenir la même réponse que 10 mg de morphine.
L’héroïne est une variante de la morphine dans laquelle les groupes hydroxyles en position C3 et C6 sont tous deux acétylés. Cette modification augmente la solubilité dans les lipides de la membrane, favorisant ainsi une pénétration rapide dans le système nerveux central. Contrairement à la codéine, les groupes acétyles sont facilement métabolisés en hydroxyles. Par conséquent, l’héroïne est environ deux fois plus puissante que la morphine.
Pour compenser la faible affinité de liaison de la codéine, des dérivés ont été étudiés, notamment l’hydrocodone. Dans ce composé, l’hydroxyle en position C6 est converti en cétone, ce qui accroît sa lipophilie tout en préservant la double liaison. Cependant, le groupe carbonyle modifie la position de l’oxygène par rapport au donneur de liaison hydrogène, tandis que la suppression de la double liaison entre les positions C3 et C4 permet de réaligner les atomes. Ces modifications ont pour effet de rendre l’hydrocodone et la morphine aussi puissantes l’une que l’autre. L’hydrocodone peut être métabolisée en un produit similaire (à l’exception de l’hydroxyle au lieu du méthoxy en C6) plus puissant, l’hydromorphone.
Une autre modification de la relation structure-activité (RSA) consiste à l’ajout d’un groupe hydroxyle en position C14 à l’hydrocodone. L’oxycodone, quant à elle, présente une biodisponibilité encore plus grande avec une puissance environ deux fois supérieure à celle de la morphine.
5.3 Résumé
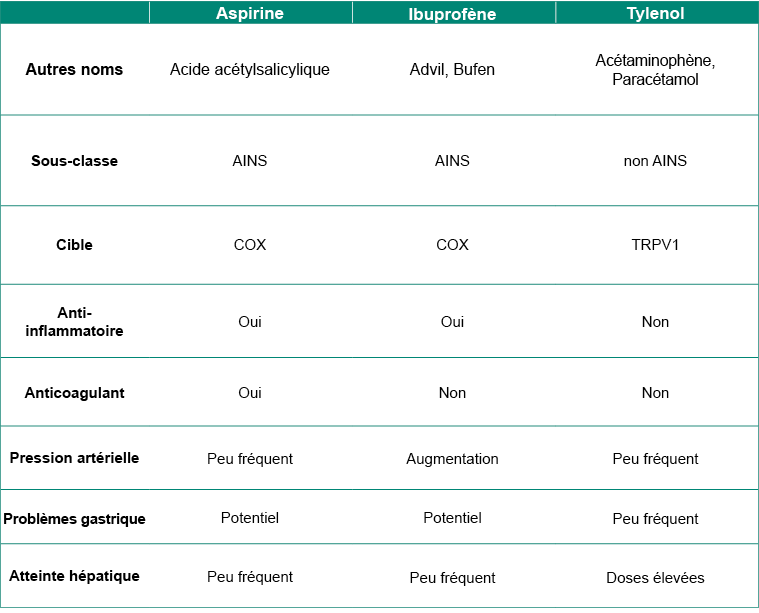
Tableau 5.1 : Résumé des médicaments analgésiques en vente libre.