4 Médicaments pour le traitement du diabète sucré
4.1 Pathologie
Le diabète sucré est une maladie qui se manifeste par une concentration élevée de glucose dans le sang. Le glucose est le principal type de glucide que l’organisme utilise comme source d’énergie. Les glucides font partie des trois macronutriments essentiels, aux côtés des protéines et des lipides. Contrairement à l’hyperlipidémie, qui se caractérise par une forte concentration de lipides hydrophobes dans le sang, la situation est quelque peu inversée dans le cas du diabète sucré. Le sang contient une concentration élevée de sucres, qui sont hautement polaires en raison des nombreux groupes hydroxyles présents sur chaque molécule de sucre. Le sang peut alors devenir très « collant » et visqueux. Ce problème devient plus grave dans les organes pourvus de vaisseaux sanguins minuscules, tels que les yeux, les reins et les autres extrémités. En effet, il devient difficile pour le sang d’atteindre ces zones et d’y transporter les nutriments essentiels ou les cellules immunitaires nécessaires. Il est donc très important de maintenir des concentrations appropriées de sucre dans le sang.
Le diabète sucré est une maladie chronique dont il existe trois différents types : diabète de type 1, diabète de type 2 et diabète gestationnel. Le diabète de type 1 est généralement considéré comme une maladie génétique, d’apparition précoce et de moindre occurrence, représentant environ 10 % des cas de diabète. Le diabète de type 2 peut également avoir une composante génétique, mais il est largement influencé par le mode de vie et se manifeste plus tardivement. Le diabète gestationnel peut survenir durant la grossesse de la patiente et disparaît généralement de manière naturelle après l’accouchement.
L’incidence du diabète au Canada a augmenté au cours des deux dernières décennies, passant de 4,7 % en 2001 à 8,1 % en 2017. Cette hausse peut être attribuée à des changements dans l’alimentation et le mode de vie. Les glucides représentent la forme d’énergie la plus accessible pour l’organisme et constituent une grande partie du régime alimentaire naturel de l’homme. Leur digestion et leur absorption par les différents organes du corps posent les bases des efforts de la chimie médicinale dans le traitement du diabète.
4.1.1 Digestion des glucides : bouche et estomac
La digestion des glucides commence dans la bouche. Bien que l’action mécanique de la mastication aide à séparer physiquement les glucides, l’enzyme amylase salivaire, de son côté, commence à hydrolyser certains des sucres complexes. Environ 5 % de la digestion des glucides s’effectue dans la bouche. Ces enzymes sont ensuite inactivées par l’acide gastrique de l’estomac. L’activité mécanique de l’estomac, combinée à l’acidité, peut favoriser une meilleure séparation des glucides. Cependant, la digestion des glucides ne s’effectue pas principalement à ce stade.
4.1.2 Digestion des glucides : intestin grêle
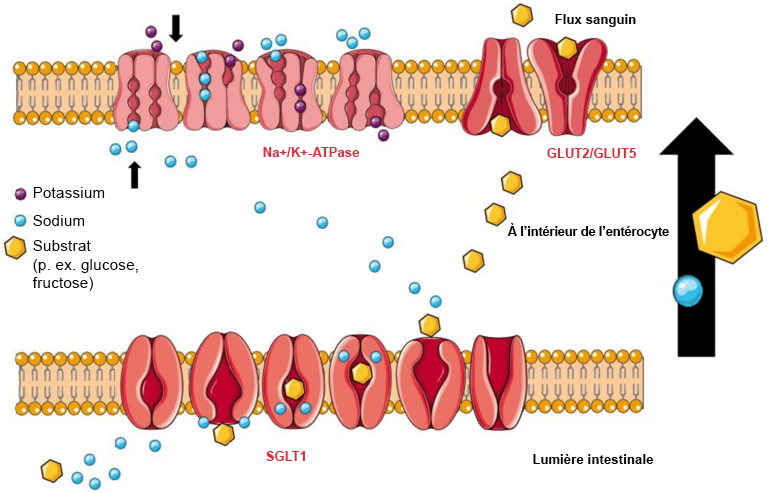
L’estomac déverse le mélange d’aliments, appelé chyme, dans l’intestin grêle, où plusieurs enzymes entrent en jeu. L’amylase pancréatique, une autre forme d’amylase, poursuit la décomposition des sucres plus complexe. Une autre enzyme, l’α-glucosidase, décompose les polymères de sucre en monomères, tels que le glucose ou le fructose. Ces monomères peuvent ensuite être absorbés grâce à différents transporteurs. Les protéines présentes dans les cellules de l’intestin grêle sont exprimées de manière asymétrique à la surface de la membrane cellulaire (Figure 4.1). Par exemple, le cotransporteur sodium-glucose type 1 (SGLT1) permet le transport du glucose dans les cellules à partir de la lumière de l’intestin grêle. Le transporteur de glucose 2 (GLUT2) est orienté vers la lumière des vaisseaux sanguins et dépose le glucose dans la circulation sanguine. Ces deux transporteurs jouent un rôle essentiel et appartiennent à une grande famille de transporteurs d’hydrates de carbone. Leur expression varie selon les tissus dans l’ensemble du corps. Le SGLT1 est l’un des douze membres de la famille des protéines SLC5A (solute carrier family member 1, ou protéine de la famille des transporteurs de solution**). Les transporteurs GLUT forment une famille de 14 protéines qui ont des affinités variables pour le glucose et les autres monomères glucidiques (par exemple, GLUT5 absorbe le fructose).
Dès que le chyme est déversé dans l’intestin grêle, il déclenche aussitôt la libération d’hormones que l’on nomme incrétines (Figure 4.2). Deux incrétines sont très importantes : le peptide-1 similaire au glucagon (GLP1) et le peptide insulinotrope dépendant du glucose (PIDG). Ces polypeptides, produits par des cellules spécifiques de l’intestin ascendant et descendant, se dirigent vers le pancréas pour l’assister dans la préparation à la montée en flèche du taux de sucre dans le sang. Ainsi, l’activité des incrétines se déclenche avant même que le glucose n’entre dans la circulation sanguine.
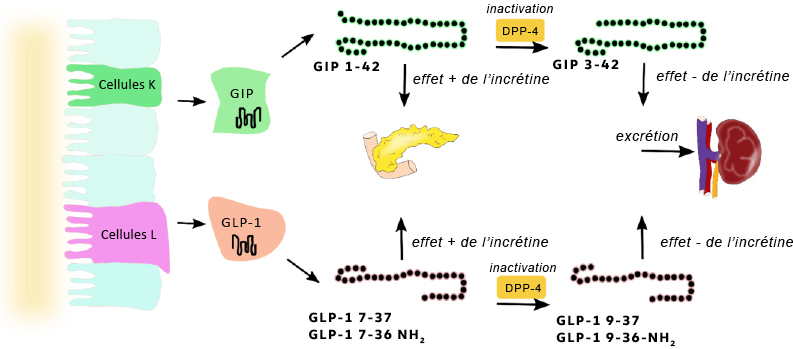
Le GIP joue de multiples rôles, notamment celui de favoriser la sécrétion d’insuline, d’inhiber la libération de glucagon et de retarder la vidange gastrique, permettant ainsi une digestion plus lente et atténuant les montées en flèche du taux glycémique. Les incrétines procurent également la sensation de satiété, ce qui réduit la consommation d’aliments. Ces hormones sont rapidement inactivées par un groupe d’enzymes appelées dipeptidyl-peptidase 4 (DDP-4) présents dans la circulation sanguine, et leur action ne persiste que pendant quelques minutes.
4.1.3 Digestion des glucides : le pancréas
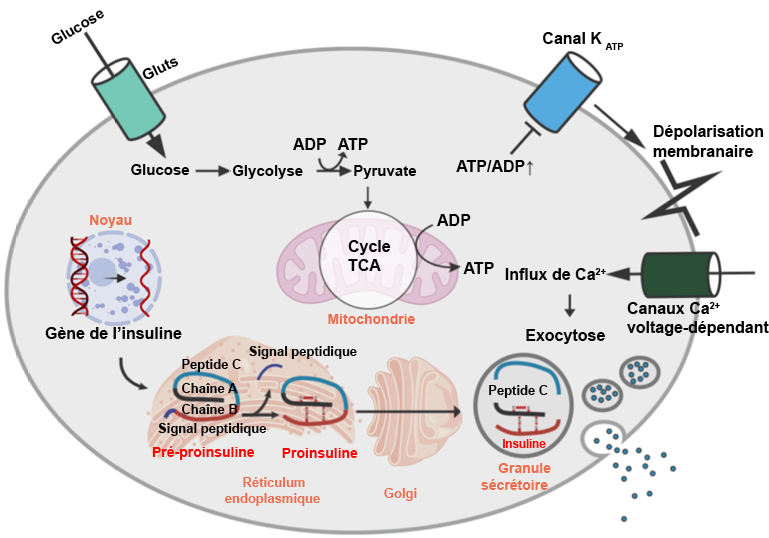
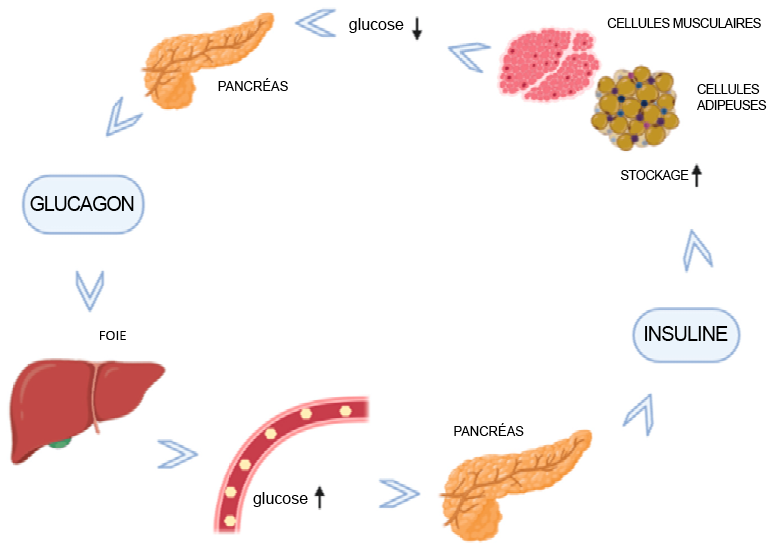
Le pancréas est l’un des organes les plus importants dans la régulation de la glycémie (Figure 4.3). Dès que le glucose entre dans la circulation sanguine au niveau de l’intestin grêle, plusieurs organes du corps entrent en action pour réguler son métabolisme. Les cellules pancréatiques disposent de transporteurs GLUT2, similaires à ceux présents dans l’intestin grêle, qui ont pour rôle d’absorber le glucose du sang, en particulier au niveau des cellules β pancréatiques. L’augmentation des concentrations intracellulaires de glucose déclenche la production d’ATP (par l’intermédiaire de la glycolyse, du cycle de Krebs, etc.), ce qui conduit à un niveau élevé d’ATP L’ATP se lie aux canaux KATP (canaux potassiques sensibles à l’ATP), ce qui provoque la fermeture de ces canaux potassiques, entraînant ainsi une modification de la polarisation de la membrane. Ce changement déclenche l’ouverture de canaux Ca2+ voltage-dépendant. L’augmentation de la teneur en ions Ca2+ déclenche la sécrétion d’insuline par les cellules β dans la circulation sanguine. L’insuline, une hormone essentielle dans la digestion des glucides, est responsable de l’augmentation de la perméabilité des cellules au glucose qui contribue à réduire les concentrations de glucose dans le sang.
L’insuline, présente dans la circulation sanguine, se lie aux récepteurs d’insuline situés sur divers types de cellules. Cette liaison déclenche une cascade de signaux intracellulaires, aboutissant à la translocation des transporteurs de glucose vers la surface cellulaire. L’activité des transporteurs de glucose GLUT4 présents dans les muscles squelettiques et cardiaques est dépendante de l’expression de l’insuline, tandis que celle des transporteurs du pancréas et de l’intestin grêle, les protéines GLUT2, est indépendante de l’insuline. La présence de ces transporteurs permet l’absorption du glucose dans le sang, fournissant ainsi une source d’énergie aux cellules et rétablissant un taux normal de glucose.
Lorsque le corps a besoin d’énergie, mais qu’il n’y a pas d’apport alimentaire, le taux de sucre dans le sang peut commencer à chuter. Les cellules α du pancréas détectent la baisse du taux de sucre et libèrent un polypeptide, le glucagon. Le glucagon, une hormone complémentaire à l’insuline, signale au foie de libérer du glucose, ce dernier étant stocké dans le foie sous la forme d’un polymère connu sous le nom de glycogène. La libération de glucose entraîne une augmentation de la glycémie, et c’est ainsi que l’interaction entre le pancréas et le foie, par l’intermédiaire des hormones insuline et glucagon, régule l’homéostasie de l’organisme (Figure 4.4).
4.1.4 Digestion des glucides : le foie
Le foie joue un rôle essentiel dans le maintien du taux de sucre dans le sang. Le foie emmagasine l’excès de glucose sous forme de glycogène, un polymère de glucose très dense. Le pancréas et le foie travaillent de concert pour s’assurer que le taux de glucose dans le sang reste constant en fonction des besoins quotidiens et des ressources dont dispose l’organisme. Par conséquent, lorsque le corps dispose d’un surplus de glucose, le pancréas sécrète de l’insuline, incitant ainsi le foie à emmagasiner du glucose. En revanche, lorsque la glycémie diminue, le pancréas libère du glucagon, stimulant le foie à dégrader le glycogène pour libérer du glucose dans la circulation sanguine.
4.2 L’insuline
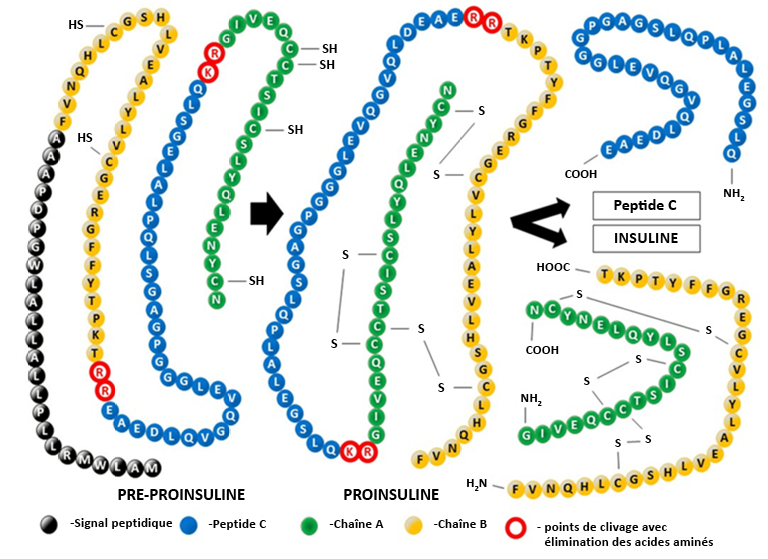
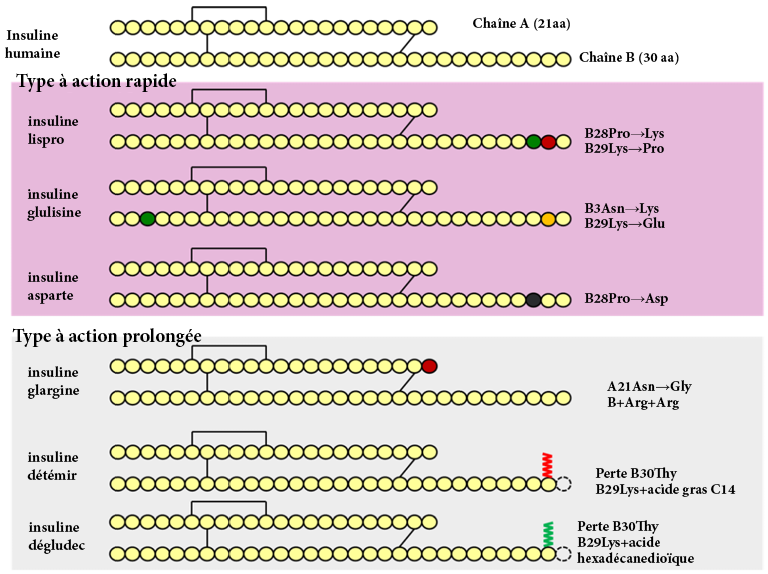
L’insuline est un polypeptide synthétisé naturellement par le pancréas. Elle est composée de deux chaînes polypeptidiques : la chaîne A (21 acides aminés) et la chaîne B (30 acides aminés). Leur poids moléculaire combiné est d’environ 6 kDa. De plus, l’insuline comporte trois liaisons disulfures essentielles :
- intramoléculaire : Cys6 (chaîne A) et Cys11 (chaîne A)
- intermoléculaire : Cys7 (chaîne A) avec Cys7 (chaîne B)
- intermoléculaire : Cys20 (chaîne A) et Cys19 (chaîne B)
L’insuline est biosynthétisée sous la forme d’une chaîne polypeptidique unique dans les cellules β du pancréas. Les ribosomes synthétisent la prépro-insuline, un polypeptide à quatre segments continus. (Figure 4.5) Le segment N-terminal contient la séquence signal pour la voie excrétrice, puis est séparé du segment protéique principal dans le réticulum endoplasmique rugueux, ce qui transforme le polypeptide en pro-insuline. Ce processus permet à la protéine de se replier sur elle-même et de former les trois ponts disulfures qui la caractérisent. Dans le réseau trans-Golgi, le peptide de la pro-insuline est clivé en deux endroits pour former la molécule d’insuline finale. Cette variante de la protéine est ensuite sécrétée par les cellules bêta du pancréas et circule dans la circulation sanguine.
La biosynthèse de l’insuline se produit dans le pancréas, où elle atteint un taux intracellulaire très élevé. De même, les cellules β maintiennent aussi une concentration relativement élevée d’ions Zn2+, ce qui, associé à la haute concentration d’insuline, entraîne la cristallisation des peptides d’insuline autour des ions Zn 2+. Les cristaux présentent alors un axe de symétrie d’ordre 3. Au centre du polymère se trouvent deux ions Zn2+, entourés par six molécules d’insuline, c’est-à-dire un trimère de dimères. Des résidus histidines donnés de la chaîne B de l’insuline coordonnent les ions Zn2+ (Figure 4.6).
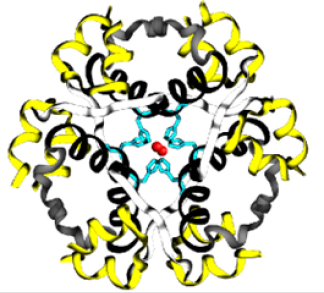
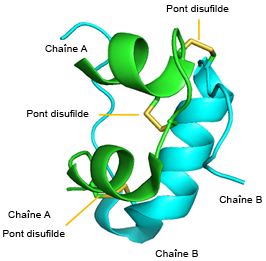
FIGURE 4.6 Les monomères d’insuline se combinent pour former des hexamères qui coordonnent un ion zinc central. Source de l’image : (Fig 1) par Beatrice Rosetti et Silvia Marchesan est utilisée sous licence CC-BY 4.0 et (Fig 2) par Harish Vashisth a été modifiée (recadrée) et utilisée sous licence CC-BY 4.0.
Ces hexamères deviennent extrêmement importants sur le plan pharmacologique. En fait, les hexamères d’insuline sont inactifs dans cet état et ne peuvent pas remplir leur fonction biologique naturelle. Cependant, lorsque le pancréas libère de l’insuline dans la circulation sanguine, la concentration de Zn2+ diminue considérablement. Les hexamères se dissocient alors rapidement en monomères, devenant ainsi actifs. Par conséquent, l’équilibre et la transition entre les hexamères d’insuline et les monomères sont critiques. Les vaisseaux cellulaires qui emmagasinent l’insuline sont aussi plutôt acides, ce qui diminue d’autant plus la solubilité de l’insuline et favorise sa structure cristalline. L’insuline a une demi-vie très courte dans le sang (environ 3 à 10 minutes) et se dégrade rapidement. Les effets de l’insuline sont donc de courte durée, mais extrêmement puissants.
4.3 Traitements du diabète de type I
Les patients atteints de diabète de type I peuvent être génétiquement limités dans leur capacité à produire suffisamment d’insuline et ne seront pas en mesure de réagir aux fluctuations de la concentration de glucose dans le sang. Une approche thérapeutique qui a vu le jour consiste à fournir de l’insuline exogène à l’organisme. Cette insuline peut être synthétiquement produite ou extraite de sources biologiques. De cette façon, les fonctions de production d’insuline des cellules β sont substituées (Figure 4.7).
4.3.1 Agents à courte durée d’action
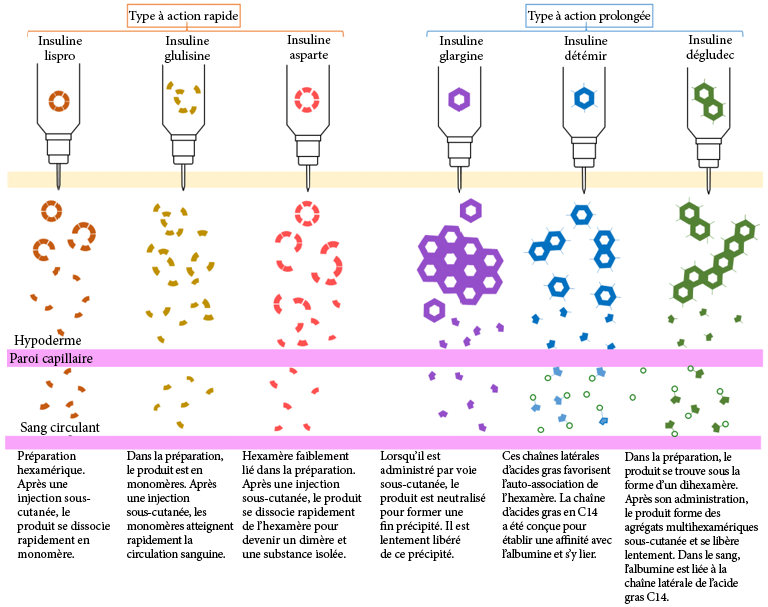
La première approche implique l’administration directe d’insuline, sous sa forme naturelle dans l’organisme. Cet agent est qualifié d’insuline régulière. L’insuline régulière partage exactement la même séquence protéique que l’insuline humaine et adopte ainsi la même structure hexamérique en présence de l’ion Zn2+. La principale différence est que l’insuline régulière est injectée par voie sous-cutanée, contrairement à l’insuline naturelle qui pénètre dans la circulation sanguine directement depuis les cellules β du pancréas. La relâche immédiate de l’insuline naturelle dans la circulation sanguine entraîne une dissociation quasi instantanée des hexamères. Lors des injections sous-cutanées, les hexamères doivent préalablement se dissocier en monomères plus petits avant de pouvoir pénétrer dans la circulation sanguine. Il en résulte que l’action de l’insuline est retardée. Avant d’être efficacement absorbée, elle doit se dissocier en monomères, ce processus pouvant prendre environ 30 à 60 minutes. Ce délai peut s’avérer incommodant, car les patients doivent attendre environ 30 minutes avant de manger à la suite d’une injection.
4.3.2 Agents à action rapide
Afin de compenser ce délai de réaction, diverses modifications ont été apportées et analysées dans la séquence peptidique de l’insuline. L’une des modifications qui a permis d’améliorer l’efficacité de l’insuline consistait à intervertir les acides aminés Pro et Lys aux positions B28 et B29. Ce changement d’acide aminé déstabilise les liaisons impliquées dans l’hexadimérisation, entraînant une dissociation rapide et favorisant ainsi une absorption immédiate après l’injection. Le profil d’absorption résultant de cette variante de l’insuline, dénommé insuline lispro, est inférieur à 15 minutes.
Une autre variante a été créée sous le nom d’insuline aspart, dans laquelle l’acide aminé Pro en position B28 a été remplacé par un Asp. L’introduction d’une charge négative perturbe l’interface de l’hexadimère, entraînant un profil d’absorption rapide, similaire à celui de l’insuline lispro. Si l’on compare ces deux agents, l’insuline lispro se caractérise par un taux d’absorption légèrement plus rapide, un pic plasmatique plus précoce et une décroissance plus rapide, tandis que l’insuline aspart offre une stabilité légèrement supérieure. Toutefois, ces différences restent mineures et, dans la pratique, l’utilisation de ces deux produits est souvent difficile à distinguer.
L’insuline glulisine est une variante de l’insuline dans laquelle la Gly en position B3 et la Pro en position B28 ont été remplacées par une Lys et une Glu, respectivement. En plus de ces modifications, la préparation contient du polysorbate 20 au lieu du Zn2+. Cette substitution est importante, car le Zn2+ joue un rôle essentiel dans le maintien de l’insuline sous forme d’hexamères. En l’absence de formation d’hexamères, le taux d’absorption est nettement plus rapide que celui de l’insuline lispro ou de l’insuline aspart.
4.3.3 Agents à action intermédiaire
Il existe des agents à action plus lente, parmi lesquels l’insuline isophane (NPH) est la plus courante. Pour celle-ci, la molécule d’insuline possède exactement la même séquence que l’insuline naturelle, mais la protamine est également ajoutée dans la préparation. La protamine, un peptide hautement chargé positivement, crée un vaste réseau d’agrégats d’hexamères d’insuline lorsqu’elle est introduite. Après leur injection, les monomères d’insuline doivent se libérer de la structure hexamérique et naviguer à travers les structures maillées présentes dans la préparation. Par conséquent, la libération de l’insuline se produit plus lentement, s’étalant sur plusieurs heures, et l’insuline NPH déploie son action plus tardivement. L’insuline NPH contribue donc à maintenir un apport régulier et modéré d’insuline dans la circulation sanguine, sans nécessiter d’injections répétées ou continues.
4.3.4 Agents à action prolongée
Certains médicaments ont également des délais d’action plus longs, ce qui retarde la libération d’insuline. Ces agents à action prolongée présentent des profils pharmacocinétiques similaires, entraînant une libération d’insuline sur une période de 12 à 24 heures. L’insuline glargine, dans laquelle une Gly remplace une Asn en position A21 et deux résidus Arg sont ajoutés à l’extrémité C-terminal de la chaîne B, est le médicament à action prolongé le plus couramment prescrit. Ces modifications réduisent le pH isoélectrique (pI) de la protéine à 6,7, ce qui entraîne sa précipitation à la fois en pH neutre et au point d’injection. Avec le temps, l’insuline glargine réintégrera lentement la solution pour se dissocier en monomères, ces derniers pouvant ensuite pénétrer dans la circulation sanguine.
Des variantes de ce type d’insuline existent également, telles que l’insuline détémir, dans laquelle une Thr en position B30 est retirée et un groupe d’acide gras (acide myristique C14) est ajouté. Cette modification crée une liaison supplémentaire avec les protéines de l’albumine. De même, l’insuline dégludec, dans laquelle une Thr en position B30 est retirée et un acide gras C16 avec un espaceur Glu est ajouté, forme des multihexamères et se lie également à l’albumine. Dans tous les cas, ces variantes introduisent des interactions supplémentaires qui prolongent le temps nécessaire à la dissociation de l’insuline en monomères actifs.
Les différents schémas de dissociation de chaque type l’insuline sont présentés dans la Figure 4.8.
4.4 Traitements du diabète de type I
Il est important de souligner que les traitements du diabète de type I nécessitent tous des injections. Ces traitements, composés de peptides de grande taille, ne conviennent pas à l’administration par voie orale. En effet, ceux-ci ne survivraient pas à un passage dans l’estomac et ne pourraient pas être absorbés par l’intestin grêle sans subir un clivage ou une modification au préalable. Le diabète de type II se caractérise généralement par une diminution de la sensibilité à l’insuline. De ce fait, l’administration d’une plus grande quantité d’insuline ne constitue pas la méthode la plus efficace pour réduire les concentrations de sucre dans le sang. Pour ces cas particuliers, un certain nombre de petites molécules ou de médicaments administrés par voie orale ont été développés.
4.4.1 Effets multiples sur la réduction de la glycémie : biguanides
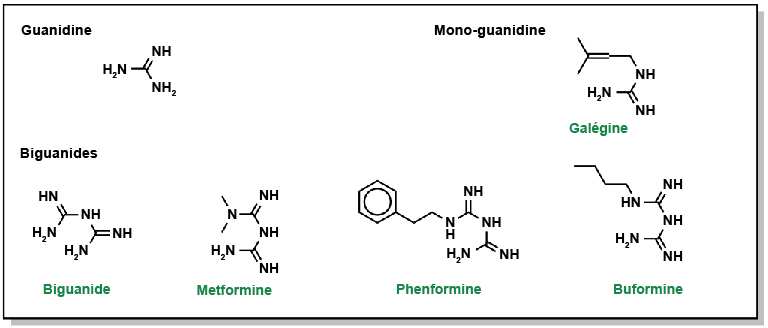
Les biguanides, découverts dans les années 1840 à partir d’herbes telles que le lilas français et riche en dérivés de guanidine, ont suscité l’intérêt. Toutefois, bon nombre de ces composés de guanidine présentaient des effets indésirables importants et n’ont pas été développés davantage. Néanmoins, dans les années 1950, ils ont été réexaminés pour le traitement du diabète, et le composé metformine a fait l’objet de recherches plus approfondies. Sur le plan de la structure chimique, un biguanide résulte de la liaison de deux molécules de guanidine. Dans le cas de la metformine, elle est dotée de deux groupes méthyle à l’une de ses extrémités. Cependant, d’autres composés, comme la butformine (avec un groupe butyle) ou la phénformine (avec un groupe phényle), ont présenté plusieurs effets secondaires et se sont avérés moins efficaces dans le traitement du diabète (Figure 4.9). La metformine est une molécule relativement plane et monoprotonée à pH neutre. Sa biodisponibilité orale est d’environ 50 % et son Tmax est d’environ 2,5 heures. Le mécanisme d’action de la metformine suscite encore de nombreux débats, mais on lui attribue au moins trois effets principaux :
- réduction de la production de glucose par le foie (bloque la gluconéogenèse)
- réduction de l’absorption intestinale du glucose
- augmentation de la sensibilité à l’insuline
Des études physiologiques et biochimiques ont révélé que la metformine pénètre dans le foie par l’intermédiaire de l’OCT1 (transporteur de cations organiques) et bloque finalement la chaîne de transport d’électrons nécessaire à la phosphorylation oxydative et à la production d’énergie (Figure 4.10). Étant donné que la molécule clé dans le transfert d’électrons et la production d’énergie, le NADPH, dépose des électrons dans la chaîne de transport d’électrons pour régénérer le NADP+, l’arrêt de cette chaîne entraîne une accumulation de NADPH dans la cellule. L’accumulation excessive de NADPH entraîne diverses réactions biochimiques et signale aux cellules qu’elles doivent cesser de convertir le glycogène en glucose. De plus, l’accumulation de NADPH peut entraîner la réduction du pyruvate par le NADPH, formant ainsi de l’acide lactique. C’est pourquoi l’acidose lactique est l’un des effets indésirables potentiels de la metformine.
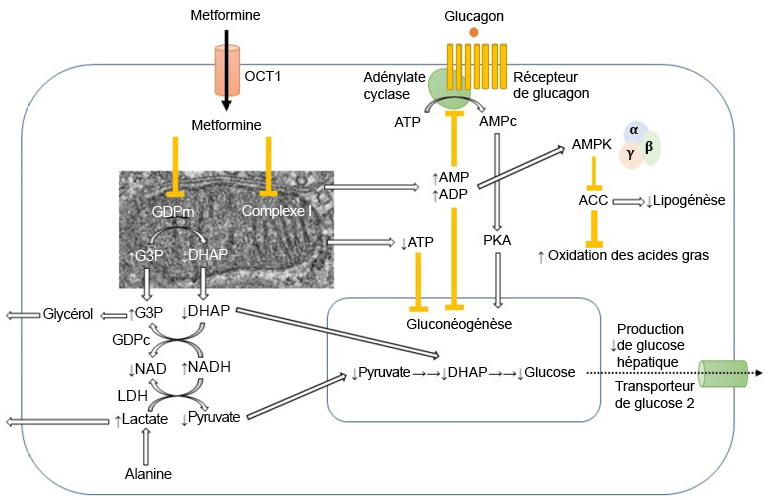 FIGURE 4.10 Différents effets de la metformine dans la cellule. Source de l’image : (Fig 1) par Ichiro Nojima et Jun Wada est utilisée sous licence CC-BY 4.0.
FIGURE 4.10 Différents effets de la metformine dans la cellule. Source de l’image : (Fig 1) par Ichiro Nojima et Jun Wada est utilisée sous licence CC-BY 4.0.
La metformine est ultimement éliminée par les reins. En raison de ses effets, elle est généralement interrompue chez un patient hospitalisé pour une affection nécessitant une tomodensitométrie, puisque l’agent de contraste est également traité par les reins. Si la metformine et l’agent de contraste sont administrés simultanément, la metformine peut persister plus longtemps dans l’organisme, entraînant des concentrations excessives et mener à une acidose lactique. En effet, ces deux substances sont en compétition pour la clairance rénale.
4.4.2 Augmentation de la libération d’insuline : sulfonylurées
Les sulfonylurées sont des médicaments qui renferment un groupe fonctionnel appelé sulfonylurée, généralement situé près d’un anneau aryle. Ces médicaments se lient aux récepteurs sulfonylurées (SUR) présents dans le pancréas (Figure 4.11). Les protéines SUR sont des transporteurs ABC qui font partie d’un plus grand complexe de canaux potassiques dépendant de l’ATP (canaux KATP) et contrôlent la libération de l’insuline. Ces médicaments sont utilisés depuis plusieurs décennies et sont relativement peu coûteux. En conditions normales, la présence de glucose dans les cellules du pancréas entraîne aussi une élévation d’ATP intracellulaire. Ces niveaux élevés d’ATP bloquent le canal KATP, provoquant ainsi une dépolarisation de la membrane qui, à son tour, déclenche l’ouverture des canaux Ca2+ voltage-dépendants et l’afflux d’ions Ca2+. Comme indiqué précédemment (Figure 4.3), l’introduction de Ca2+ entraîne la libération d’insuline.
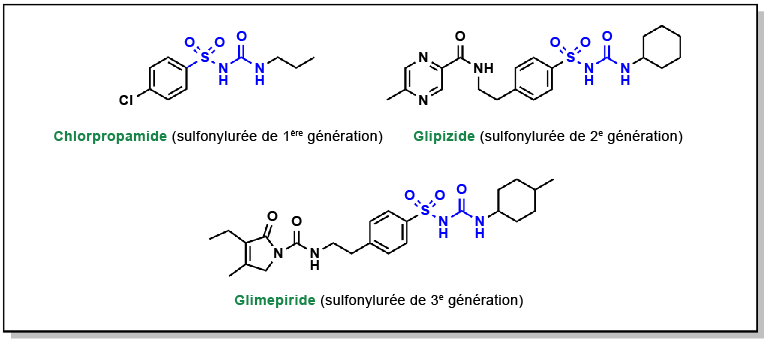
L’administration de sulfonylurées produit une réponse identique à celle d’un niveau élevé d’ATP intracellulaire. Plus précisément, la sulfonylurée se lie au canal KATP, entraînant la dépolarisation de la membrane et l’afflux de Ca2+. Cela dissocie essentiellement la libération d’insuline des concentrations de glucose sanguin. Cependant, il est important de souligner que l’administration excessive de sulfonylurées peut provoquer une baisse significative de la glycémie.
La prise de poids est un autre des effets secondaires causés par les sulfonylurées. En effet, ces médicaments stimulent une libération accrue d’insuline, ce qui incite les cellules de l’organisme à absorber davantage de glucose. De plus, les canaux KATP sont également présents dans d’autres tissus de l’organisme, notamment le cœur. La liaison non spécifique des sulfonylurées à ces canaux peut accroître le risque d’événements cardiaques.
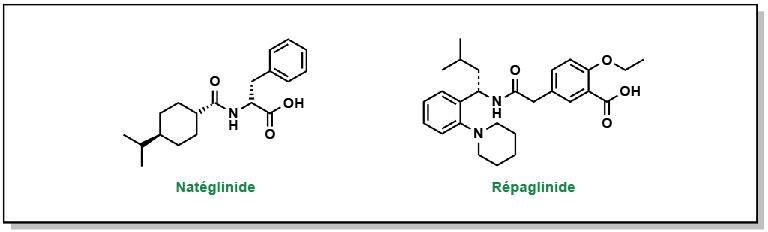
4.4.3 Augmentation de la libération d’insuline : méglitinides
Les méglitinides fonctionnent de manière similaire aux sulfonylurées : elles se lient au canal KATP, mais à un site différent de celui où se fixent les sulfonylurées sur la protéine SUR (Figure 4.12). Cependant, l’interaction entre les méglitinides et la protéine SUR n’est pas aussi forte que la liaison sulfonylurée-SUR. Il en résulte un effet relativement plus faible et une durée d’action plus courte. Cette plus faible activité peut s’avérer très utile, car elle offre une plus grande flexibilité dans les choix thérapeutiques et les méglitinides ne sont pas associés à des événements cardiovasculaires indésirables. Les méglitinides offrent une alternative intéressante lorsque le patient présente une allergie aux sulfonylurées.
4.4.4 Augmentation de la production d’insuline : inhibiteurs de la dipeptidyl-peptidase 4 (DPP-4)
Outre les hormones produites par le pancréas, il existe également des hormones produites par l’intestin grêle appelées incrétines, notamment GIP et GLP1. Ces incrétines stimulent le corps pour le préparer à l’ingestion de nourriture ainsi que pour la production et la libération d’insuline. Ces hormones sont très rapidement dégradées par l’enzyme dipeptidyl peptidase 4 (DDP-4). Prolonger les effets du GIP et du GLP1 (par l’inhibition de la DDP-4 avec de petites molécules) peut accroître les taux d’insuline dans l’organisme, tout en ayant d’autres effets sur la digestion et l’absorption. Les inhibiteurs de la DPP-4 se lient au site d’interaction du GLP1 avec la protéine DPP-4 et sont catégorisés comme des inhibiteurs d’interaction protéine-protéine. Plusieurs médicaments contiennent des azolopyrimidines qui jouent un rôle essentiel dans les liaisons hydrogène au niveau du site de liaison (Figure 4.13).
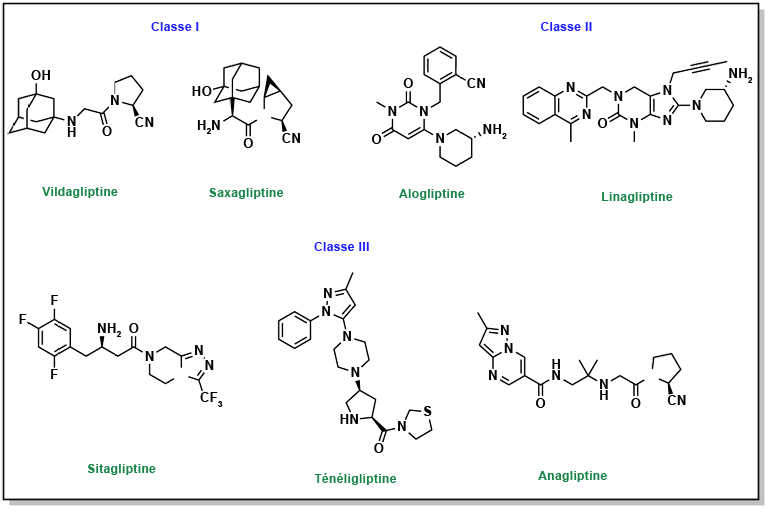
L’un des effets secondaires courants de ces médicaments est la perte de poids, bien qu’ils stimulent également la libération d’insuline, à l’instar des sulfonylurées ou des méglitinides. En effet, la durée de vie prolongée des incrétines entraîne une plus grande sensation de satiété, ce qui réduit généralement l’appétit et, par conséquent, l’apport calorique.
4.4.5 Augmentation de la production d’insuline : agonistes de GLP1
Les agonistes du GLP1, tout comme les inhibiteurs de la DPP-4, nécessitent une injection directe de l’incrétine GLP1 (similaire à l’administration d’insuline dans le cas du diabète de type 1). Le GLP1 est privilégié par rapport au GIP en raison de ses effets plus puissants. Ces médicaments reposent sur deux structures peptidiques basées sur le GLP1 et l’exendine-4 (initialement isolée à partir du venin d’un reptile nommé Monstre de Gila). Étant donné qu’il ne s’agit pas d’une incrétine de mammifère, l’exendine-4 n’est pas un substrat de la DPP-4 et possède une durée de vie plus longue. Tout comme les inhibiteurs de la DPP-4, ces médicaments ralentissent la digestion des aliments, augmentent la production d’insuline et renforcent la sensation de satiété.

4.4.6 Blocage de l’absorption du glucose : inhibiteurs de l’α-Glucosidase
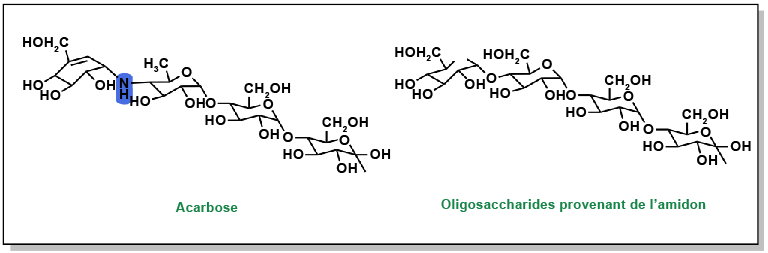
L’absorption des glucides se produit dans l’intestin grêle. Bloquer cette absorption peut donc servir de mesure préventive à la réduction du taux de glucose dans le sang. Les oligosaccharides de grande taille ne peuvent pas être absorbés directement et doivent être clivés en monosaccharides. Ce processus s’effectue grâce à l’action des α-glucosidases présentes au niveau de la bordure en brosse de l’intestin grêle. Le médicament acarbose présente une structure très similaire à celle des oligosaccharides, mais il a une affinité nettement plus élevée avec l’α-glucosidase et ne peut pas être clivé. Il entraîne un blocage compétitif du site actif de l’α-glucosidase, empêchant ainsi le clivage des oligosaccharides en monosaccharides et bloquant leur absorption. Ces inhibiteurs peuvent se révéler plus efficaces que les sulfonylurées traditionnelles, car ils ont pour objectif de prévenir activement les montées en flèche de glycémie, plutôt que de traiter les taux élevés après l’ingestion de nourriture.
4.4.7 Blocage de la réabsorption du glucose : inhibiteurs de SGLT-2
Les patients atteints de diabète de type 2 présentent une concentration accrue de glucose dans le sang, ce qui se traduit également par des niveaux plus élevés de glucose dans les reins. Les néphrons des reins sont conçus pour réabsorber le glucose par l’intermédiaire de cotransporteurs sodium-glucose, principalement le SGLT-2) (Figure 4.16). Cette fonction est utile d’un point de vue physiologique puisqu’elle permet de retenir les nutriments. Toutefois, le blocage de ces transporteurs chez les patients diabétiques de type II peut réduire la glycémie.
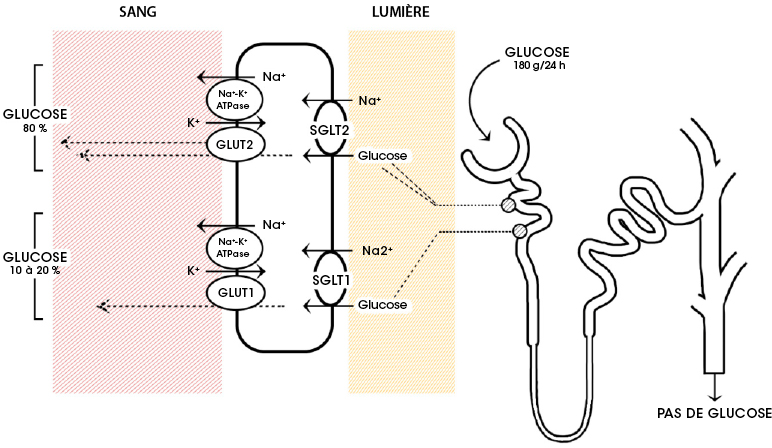 FIGURE 4.16 Réabsorption du glucose par le néphron du rein. Source de l’image : (Fig 1) par Daria M. Keller, Natasha Ahmed, Hamza Tariq, Malsha Walgamage, Thilini Walgamage, Azad Mohammed, Jadzia Tin-Tsen Chou, Marta Kałużna-Oleksy, Maciej Lesiak et Ewa Straburzyńska-Migaj est utilisée sous licence CC-BY 4.0.
FIGURE 4.16 Réabsorption du glucose par le néphron du rein. Source de l’image : (Fig 1) par Daria M. Keller, Natasha Ahmed, Hamza Tariq, Malsha Walgamage, Thilini Walgamage, Azad Mohammed, Jadzia Tin-Tsen Chou, Marta Kałużna-Oleksy, Maciej Lesiak et Ewa Straburzyńska-Migaj est utilisée sous licence CC-BY 4.0.
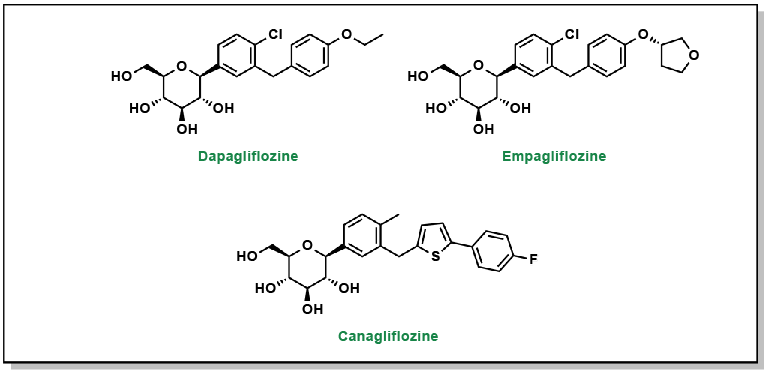
La phlorizine est le premier médicament à cet effet et se compose d’un fragment de glucose lié à un système de deux cycles phényles (la partie « aglycone ») reliés par un pont éthylène (Figure 4.17).
4.4.8 Réduction indirecte de la glycémie : blocage des triglycérides avec les thiazolidinediones/glitazones
Les thiazolidinediones, également dénommées glitazones, stimulent les cellules à utiliser les glucides/glucose comme principale source d’énergie, ce qui abaisse le taux de glucose sanguin (Figure 4.18).
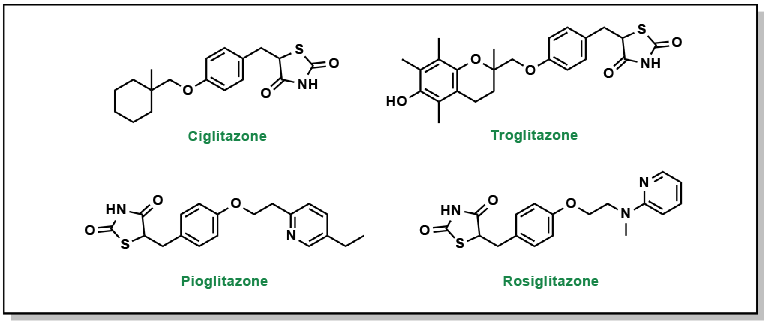 FIGURE 4.18 Structures des thiazolidinediones.
FIGURE 4.18 Structures des thiazolidinediones.
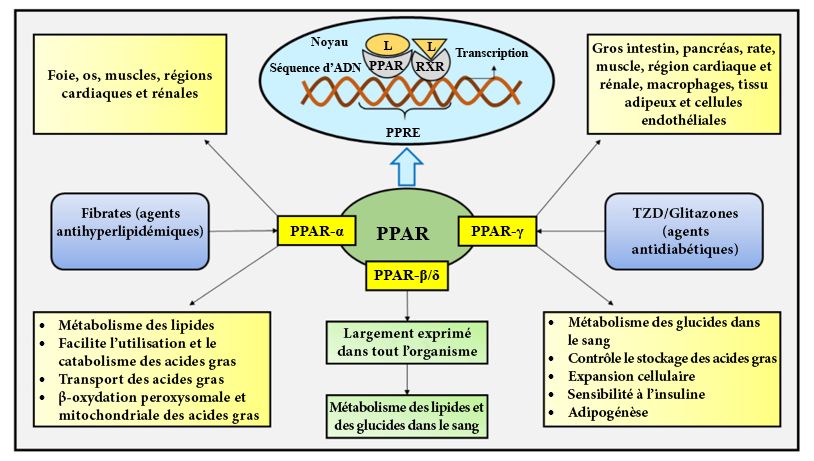
Ces médicaments sont des agonistes des proliférateurs de péroxysomes de type gamma (PPARγ), un facteur de transcription qui forme un complexe avec le récepteur rétinoïde X (RXR). Il en résulte une augmentation de l’expression des gènes responsables du stockage des acides gras, réduisant ainsi la quantité d’acides gras libres dans la circulation sanguine et favorisent une dépendance accrue des cellules au glucose. GLUT4 est une autre cible génétique des PPARγ, contribuant à l’augmentation de la quantité de glucose dans les cellules (Figure 4.19). Cependant, ce stockage plus important des acides gras est aussi corrélé à une réduction de la formation d’ostéoblastes. De ce fait, l’utilisation prolongée des thiazolidinediones entraîne une diminution de la densité minérale osseuse et accroît le risque de fractures.
4.5 Résumé
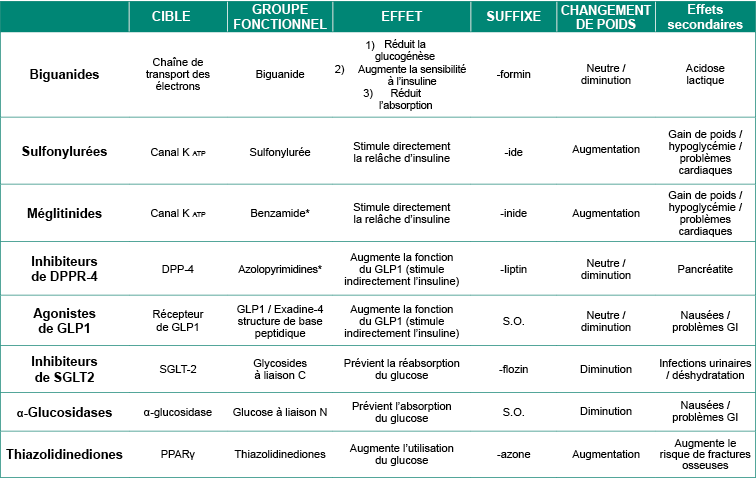
Le tableau suivant résume les traitements du diabète de type II. Les différents types d’insulines utilisés pour le traitement du diabète de type I sont résumés dans la Figure 4.8.
Tableau 4.1 Propriétés pharmacologiques des traitements du diabète de type II.